

Genome-Wide Association Study of Body Size Traits in Luning Chickens Using Whole-Genome Sequencing
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotypes
2.2. Sequencing, Quality Control, and Annotation
2.3. Statistical Analysis
3. Results
3.1. Phenotypic Statistics, Population Structure, and SNP and INDEL Characteristics
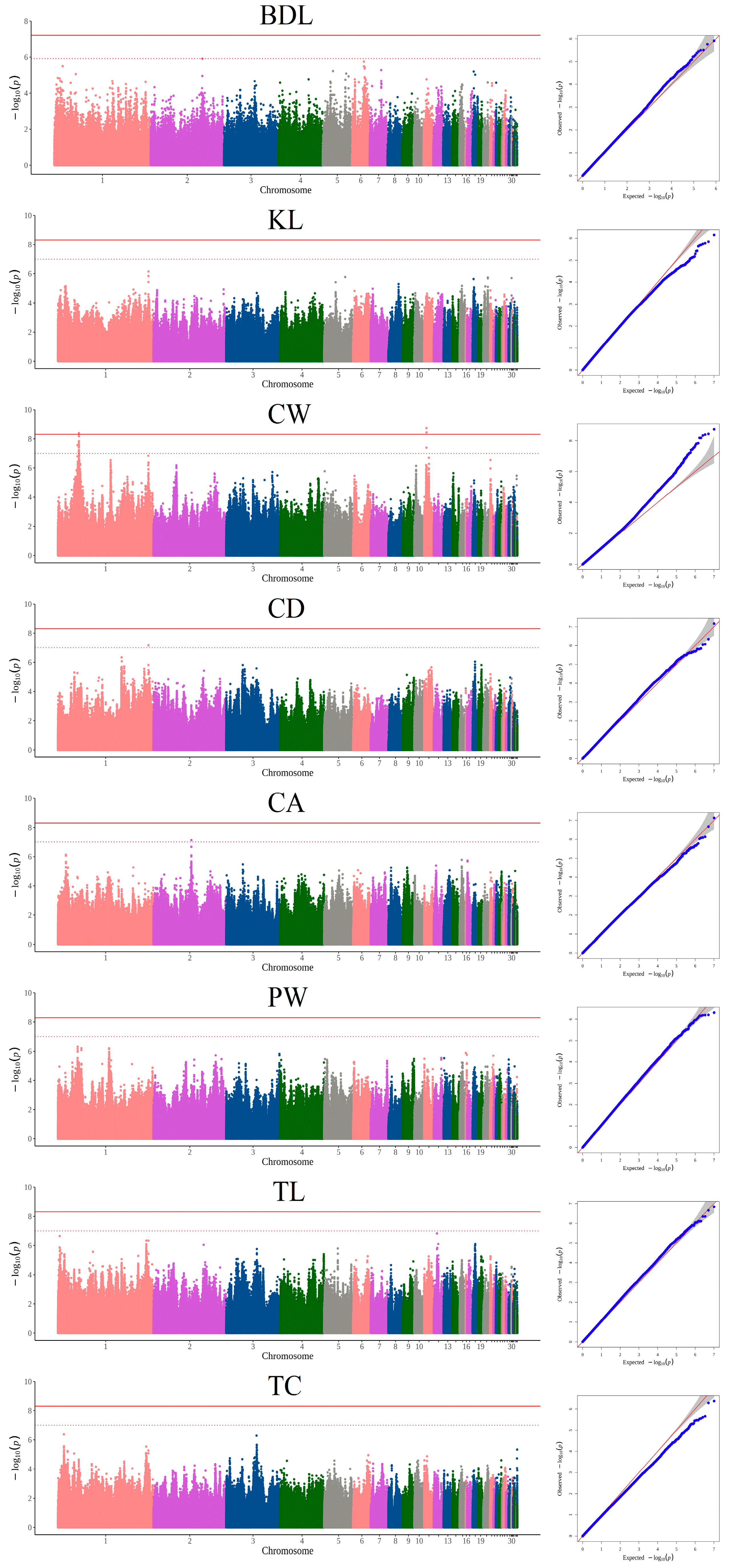
3.2. GWAS Based on SNPs and Candidate Genes
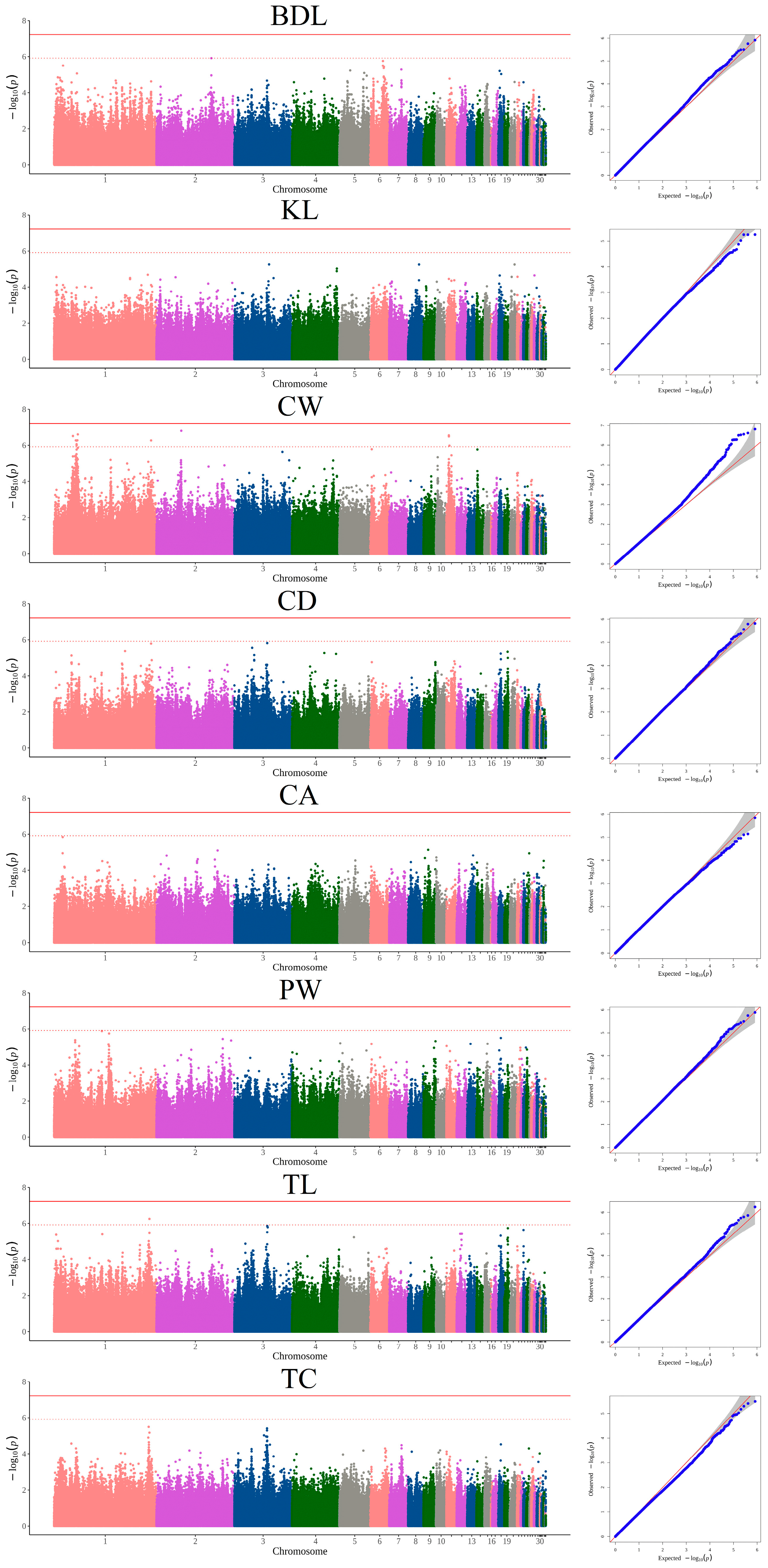
3.3. GWAS Based on INDELs and Candidate Genes
3.4. Linkage Disequilibrium (LD) Block Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FAO. World Food and Agriculture Statistical Yearbook 2023; FAO: Rome, Italy, 2023. [Google Scholar]
- Tavárez, M.A.; Solis de los Santos, F. Impact of genetics and breeding on broiler production performance: A look into the past, present, and future of the industry. Anim. Front. 2016, 6, 37–41. [Google Scholar] [CrossRef]
- Yang, R.; Xu, Z.; Wang, Q.; Zhu, D.; Bian, C.; Ren, J.; Huang, Z.; Zhu, X.; Tian, Z.; Wang, Y.; et al. Genome-wide association study and genomic prediction for growth traits in yellow-plumage chicken using genotyping-by-sequencing. Genet. Sel. Evol. GSE 2021, 53, 82. [Google Scholar] [PubMed]
- Sheng, Z.; Pettersson, M.E.; Hu, X.; Luo, C.; Qu, H.; Shu, D.; Shen, X.; Carlborg, O.; Li, N. Genetic dissection of growth traits in a Chinese indigenous × commercial broiler chicken cross. BMC Genom. 2013, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Peng, D.; Gu, X.; Gong, Y.; Sheng, Z.; Hu, X. Polygenic Basis and Variable Genetic Architectures Contribute to the Complex Nature of Body Weight—A Genome-Wide Study in Four Chinese Indigenous Chicken Breeds. Front. Genet. 2018, 9, 229. [Google Scholar]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar]
- Ding, R.; Zhuang, Z.; Qiu, Y.; Wang, X.; Wu, J.; Zhou, S.; Ruan, D.; Xu, C.; Hong, L.; Gu, T.; et al. A composite strategy of genome-wide association study and copy number variation analysis for carcass traits in a Duroc pig population. BMC Genom. 2022, 23, 590. [Google Scholar]
- Manzanilla-Pech, C.I.V.; Difford, G.F.; Sahana, G.; Romé, H.; Løvendahl, P.; Lassen, J. Genome-wide association study for methane emission traits in Danish Holstein cattle. J. Dairy Sci. 2022, 105, 1357–1368. [Google Scholar]
- Zhao, H.; Zhu, S.; Guo, T.; Han, M.; Chen, B.; Qiao, G.; Wu, Y.; Yuan, C.; Liu, J.; Lu, Z.; et al. Whole-genome re-sequencing association study on yearling wool traits in Chinese fine-wool sheep. J. Anim. Sci. 2021, 99, skab210. [Google Scholar] [CrossRef]
- Mucha, S.; Mrode, R.; Coffey, M.; Kizilaslan, M.; Desire, S.; Conington, J. Genome-wide association study of conformation and milk yield in mixed-breed dairy goats. J. Dairy Sci. 2018, 101, 2213–2225. [Google Scholar]
- Li, Z.; Xu, Y.; Lin, Y. Transcriptome analyses reveal genes of alternative splicing associated with muscle development in chickens. Gene 2018, 676, 146–155. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [PubMed]
- Ning, C.; Wang, D.; Zhou, L.; Wei, J.; Liu, Y.; Kang, H.; Zhang, S.; Zhou, X.; Xu, S.; Liu, J.-F. Efficient multivariate analysis algorithms for longitudinal genome-wide association studies. Bioinformatics 2019, 35, 4879–4885. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar]
- Bland, J.M.; Altman, D.G. Multiple significance tests: The Bonferroni method. Br. Med. J. Clin. Res. Ed. 1995, 310, 170. [Google Scholar]
- Paaby, A.B.; Rockman, M.V. The many faces of pleiotropy. Trends Genet. TIG 2013, 29, 66–73. [Google Scholar]
- Stephens, M. A unified framework for association analysis with multiple related phenotypes. PLoS ONE 2013, 8, e65245. [Google Scholar]
- Wang, D.; Teng, J.; Zhao, C.; Zhang, X.; Tang, H.; Fan, X.; Xu, S.; Zhang, Q.; Ning, C. An Improved Linear Mixed Model for Multivariate Genome-Wide Association Studies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Spangler, S.A.; Hoogenraad, C.C. Liprin-alpha proteins: Scaffold molecules for synapse maturation. Biochem. Soc. Trans. 2007, 35 Pt 5, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, J.; Li, J.; Bao, H. Identification of candidate genes associated with slaughter traits in F2 chicken population using genome-wide association study. Anim. Genet. 2021, 52, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Pandiella, A. Membrane-anchored growth factors. Annu. Rev. Biochem. 1993, 62, 515–541. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- An, X.P.; Hou, J.X.; Li, G.; Song, Y.X.; Wang, J.G.; Chen, Q.J.; Cui, Y.H.; Wang, Y.F.; Cao, B.Y. Polymorphism identification in the goat KITLG gene and association analysis with litter size. Anim. Genet. 2012, 43, 104–107. [Google Scholar] [CrossRef]
- An, X.P.; Hou, J.X.; Gao, T.Y.; Lei, Y.N.; Song, Y.X.; Wang, J.G.; Cao, B.Y. Association analysis between variants in KITLG gene and litter size in goats. Gene 2015, 558, 126–130. [Google Scholar] [CrossRef]
- Zhao, X.; Nie, C.; Zhang, J.; Li, X.; Zhu, T.; Guan, Z.; Chen, Y.; Wang, L.; Xue, Z.L.; Yang, W.; et al. Identification of candidate genomic regions for chicken egg number traits based on genome-wide association study. BMC Genom. 2021, 22, 610. [Google Scholar] [CrossRef]
- Tan, Y.G.; Xu, X.L.; Cao, H.Y.; Zhou, W.; Yin, Z.Z. Effect of age at first egg on reproduction performance and characterization of the hypothalamo-pituitary-gonadal axis in chickens. Poult. Sci. 2021, 100, 101325. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Zhou, J.; Li, J.; Zhao, X.; Zheng, L.; Bao, H.; Wu, C. Genome-Wide Association Study Identifies Candidate Genes for Stripe Pattern Feather Color of Rhode Island Red Chicks. Genes 2022, 13, 1511. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, P.; Fu, W.; Dong, T.; Qi, C.; Liu, L.; Guo, G.; Li, C.; Cui, X.; Zhang, S.; et al. Genome-wide association study for pigmentation traits in Chinese Holstein population. Anim. Genet. 2014, 45, 740–744. [Google Scholar] [PubMed]
- Yang, Z.; Shi, H.; Ma, P.; Zhao, S.; Kong, Q.; Bian, T.; Gong, C.; Zhao, Q.; Liu, Y.; Qi, X.; et al. Darwinian Positive Selection on the Pleiotropic Effects of KITLG Explain Skin Pigmentation and Winter Temperature Adaptation in Eurasians. Mol. Biol. Evol. 2018, 35, 2272–2283. [Google Scholar] [PubMed]
- Arkell, R.S.; Dickinson, R.J.; Squires, M.; Hayat, S.; Keyse, S.M.; Cook, S.J. DUSP6/MKP-3 inactivates ERK1/2 but fails to bind and inactivate ERK5. Cell. Signal. 2008, 20, 836–843. [Google Scholar] [PubMed]
- Eblaghie, M.C.; Lunn, J.S.; Dickinson, R.J.; Münsterberg, A.E.; Sanz-Ezquerro, J.J.; Farrell, E.R.; Mathers, J.; Keyse, S.M.; Storey, K.; Tickle, C. Negative feedback regulation of FGF signaling levels by Pyst1/MKP3 in chick embryos. Curr. Biol. 2003, 13, 1009–1018. [Google Scholar] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Vo, A.H.; Swaggart, K.A.; Woo, A.; Gao, Q.Q.; Demonbreun, A.R.; Fallon, K.S.; Quattrocelli, M.; Hadhazy, M.; Page, P.G.; Chen, Z.; et al. Dusp6 is a genetic modifier of growth through enhanced ERK activity. Hum. Mol. Genet. 2019, 28, 279–289. [Google Scholar]
- Li, C.; Scott, D.A.; Hatch, E.; Tian, X.; Mansour, S.L. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development 2007, 134, 167–176. [Google Scholar]
- Zhang, B.; Yuan, P.; Xu, G.; Chen, Z.; Li, Z.; Ye, H.; Wang, J.; Shi, P.; Sun, X. DUSP6 expression is associated with osteoporosis through the regulation of osteoclast differentiation via ERK2/Smad2 signaling. Cell Death Dis. 2021, 12, 825. [Google Scholar]
- Yuan, S.H.; Qiu, Z.; Ghosh, A. TOX3 regulates calcium-dependent transcription in neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 2909–2914. [Google Scholar]
- Liu, C.; Zheng, Y.; Hu, S.; Liang, X.; Li, Y.; Yu, Z.; Liu, Y.; Bian, Y.; Man, Y.; Zhao, S.; et al. TOX3 deficiency mitigates hyperglycemia by suppressing hepatic gluconeogenesis through FoxO1. Metabolism 2024, 152, 155766. [Google Scholar]
- Jiang, C.; Zhang, J.; Song, Y.; Song, X.; Wu, H.; Jiao, R.; Li, L.; Zhang, G.; Wei, D. FOXO1 regulates bovine skeletal muscle cells differentiation by targeting MYH3. Int. J. Biol. Macromol. 2024, 260 Pt 2, 129643. [Google Scholar] [CrossRef] [PubMed]
- Oyabu, M.; Takigawa, K.; Mizutani, S.; Hatazawa, Y.; Fujita, M.; Ohira, Y.; Sugimoto, T.; Suzuki, O.; Tsuchiya, K.; Suganami, T.; et al. FOXO1 cooperates with C/EBPδ and ATF4 to regulate skeletal muscle atrophy transcriptional program during fasting. FASEB J. 2022, 36, e22152. [Google Scholar] [CrossRef] [PubMed]
- Sharan, K.; Lewis, K.; Furukawa, T.; Yadav, V.K. Regulation of bone mass through pineal-derived melatonin-MT2 receptor pathway. J. Pineal Res. 2017, 63, e12423. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Kim, Y.A.; Lee, J.; Lee, S.; Kim, J.; Lee, S. Fat3 regulates neural progenitor cells by promoting Yap activity during spinal cord development. Sci. Rep. 2022, 12, 14726. [Google Scholar] [CrossRef] [PubMed]
- Watt, K.I.; Goodman, C.A.; Hornberger, T.A.; Gregorevic, P. The Hippo Signaling Pathway in the Regulation of Skeletal Muscle Mass and Function. Exerc. Sport. Sci. Rev. 2018, 46, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Botsch, J.J.; Junker, R.; Sorgenfrei, M.; Ogger, P.P.; Stier, L.; von Gronau, S.; Murray, P.J.; Seeger, M.A.; Schulman, B.A.; Bräuning, B. Doa10/MARCH6 architecture interconnects E3 ligase activity with lipid-binding transmembrane channel to regulate SQLE. Nat. Commun. 2024, 15, 410. [Google Scholar] [CrossRef]
- Li, Q.; Wang, N.; Du, Z.; Hu, X.; Chen, L.; Fei, J.; Wang, Y.; Li, N. Gastrocnemius transcriptome analysis reveals domestication induced gene expression changes between wild and domestic chickens. Genomics 2012, 100, 314–319. [Google Scholar] [CrossRef]
- Wang, K.S.; Liu, X.; Zheng, S.; Zeng, M.; Pan, Y.; Callahan, K. A novel locus for body mass index on 5p15.2: A meta-analysis of two genome-wide association studies. Gene 2012, 500, 80–84. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, L.; Gao, H.; Wu, Y.; Gao, X.; Zhang, L.; Zhu, B.; Song, Y.; Bao, J.; Li, J.; et al. Detection of candidate genes for growth and carcass traits using genome-wide association strategy in Chinese Simmental beef cattle. Anim. Prod. Sci. 2018, 58, 224–233. [Google Scholar] [CrossRef]
- Esmaeili-Fard, S.M.; Gholizadeh, M.; Hafezian, S.H.; Abdollahi-Arpanahi, R. Genes and Pathways Affecting Sheep Productivity Traits: Genetic Parameters, Genome-Wide Association Mapping, and Pathway Enrichment Analysis. Front. Genet. 2021, 12, 710613. [Google Scholar] [CrossRef]
- Derks, M.F.L.; Lopes, M.S.; Bosse, M.; Madsen, O.; Dibbits, B.; Harlizius, B.; Groenen, M.A.M.; Megens, H.J. Balancing selection on a recessive lethal deletion with pleiotropic effects on two neighboring genes in the porcine genome. PLoS Genet. 2018, 14, e1007661. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, J.; Wang, J.; Zhang, L.; Xu, L.; Chen, Y.; Zhu, B.; Wang, Z.; Gao, H.; Li, J.; et al. Genome-Wide Detection of Copy Number Variations and Their Potential Association with Carcass and Meat Quality Traits in Pingliang Red Cattle. Int. J. Mol. Sci. 2024, 25, 5626. [Google Scholar] [CrossRef]
- Liu, J.L.; Liang, X.H.; Su, R.W.; Lei, W.; Jia, B.; Feng, X.H.; Li, Z.X.; Yang, Z.M. Combined analysis of microRNome and 3′-UTRome reveals a species-specific regulation of progesterone receptor expression in the endometrium of rhesus monkey. J. Biol. Chem. 2012, 287, 13899–13910. [Google Scholar] [PubMed]
- Wang, X.; Bai, X.; Yan, Z.; Guo, X.; Zhang, Y. The lncRNA TUG1 promotes cell growth and migration in colorectal cancer via the TUG1-miR-145-5p-TRPC6 pathway. Biochem. Cell Biol. Biochim. Biol. Cell. 2021, 99, 249–260. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Trait | Number | Mean | SE | Max | Min | Median | CV (%) |
|---|---|---|---|---|---|---|---|
| BDL | 248 | 21.94 | 0.12 | 25.40 | 17.30 | 22.00 | 8.05 |
| KL | 247 | 12.32 | 0.09 | 15.50 | 8.50 | 12.30 | 10.37 |
| CW | 242 | 7.47 | 0.05 | 9.35 | 5.55 | 7.52 | 9.21 |
| CD | 248 | 10.88 | 0.06 | 13.20 | 8.84 | 10.80 | 8.33 |
| CA | 240 | 45.00 | 1.15 | 85.00 | 11.00 | 45.00 | 29.14 |
| PW | 248 | 6.77 | 0.06 | 9.09 | 4.50 | 6.73 | 11.68 |
| TL | 248 | 9.57 | 0.05 | 12.30 | 7.36 | 9.45 | 11.68 |
| TC | 248 | 4.57 | 0.03 | 6.00 | 3.20 | 4.60 | 11.26 |
| Trait | Chromosome | Position (bp) | Alleles | p-Value | Annotation | Gene Name |
|---|---|---|---|---|---|---|
| CW | 1 | 40,298,901 | G/A | 2.70 × 10−8 | Intronic | PPFIA2 |
| 1 | 43,072,056 | C/T | 6.24 × 10−8 | Intronic | KITLG | |
| 1 | 43,073,484 | A/G | 6.06 × 10−8 | Intronic | KITLG | |
| 1 | 43,075,104 | C/T | 3.09 × 10−8 | Intronic | KITLG | |
| 1 | 43,077,685 | C/T | 4.05 × 10−9 | Intronic | KITLG | |
| 1 | 43,079,649 | T/C | 8.08 × 10−8 | Intronic | KITLG | |
| 1 | 43,080,517 | A/C | 1.51 × 10−8 | Intronic | KITLG | |
| 1 | 43,081,408 | G/C | 4.61 × 10−9 | Intronic | KITLG | |
| 1 | 43,083,179 | C/A | 2.61 × 10−8 | Intronic | KITLG | |
| 1 | 43,084,274 | G/T | 7.15 × 10−8 | Intronic | KITLG | |
| 1 | 43,084,312 | A/G | 7.98 × 10−8 | Intronic | KITLG | |
| 1 | 43,084,631 | T/C | 3.48 × 10−8 | Intronic | KITLG | |
| 1 | 43,084,657 | T/C | 1.61 × 10−8 | Intronic | KITLG | |
| 1 | 43,084,806 | G/A | 2.02 × 10−8 | Intronic | KITLG | |
| 1 | 43,085,497 | T/C | 3.87 × 10−8 | Intronic | KITLG | |
| 1 | 43,202,979 | A/G | 6.65 × 10−8 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,207,402 | C/G | 8.53 × 10−8 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,207,404 | C/T | 8.53 × 10−8 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,207,758 | T/C | 6.56 × 10−9 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,207,760 | A/G | 6.56 × 10−9 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,208,364 | G/T | 6.44 × 10−8 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,208,452 | A/T | 1.45 × 10−8 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,208,537 | G/A | 3.74 × 10−8 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,219,314 | C/T | 5.83 × 10−8 | Intergenic | KITLG, DUSP6 | |
| 11 | 4,751,386 | A/T | 1.85 × 10−9 | Intronic | TOX3 | |
| 11 | 4,751,411 | C/T | 3.65 × 10−9 | Intronic | TOX3 | |
| 11 | 4,751,668 | A/C | 3.96 × 10−8 | Intronic | TOX3 | |
| CD | 1 | 186,174,672 | G/A | 6.82 × 10−8 | Intergenic | MTNR1B, FAT3 |
| CA | 2 | 77,942,520 | C/T | 7.43 × 10−8 | Exonic | MARCH6 |
| Trait | Chromosome | Position (bp) | Alleles | p-Value | Annotation | Gene Name |
|---|---|---|---|---|---|---|
| CW | 1 | 36,193,064 | G/GCA | 2.96 × 10−7 | Intergenic | PTPRR, TSPAN8 |
| 1 | 41,770,569 | GAGAA/G | 5.15 × 10−7 | Intergenic | SLC6A15, TSPAN19 | |
| 1 | 43,198,933 | T/TA | 8.75 × 10−7 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,206,303 | C/CACT | 9.98 × 10−7 | Intergenic | KITLG, DUSP6 | |
| 1 | 43,376,793 | C/CGTGGATCTGA | 5.37 × 10−7 | Intergenic | DUSP6, POC1B | |
| 1 | 44,656,419 | AT/A | 5.11 × 10−7 | Intronic | PLEKHG7 | |
| 1 | 45,559,065 | AT/A | 2.39 × 10−7 | Intronic | VEZT | |
| 1 | 186,174,628 | A/AGGCAGCT | 5.22 × 10−7 | Intergenic | MTNR1B, FAT3 | |
| 2 | 47,680,889 | AAC/A | 1.52 × 10−7 | Intronic | BBS9 | |
| 11 | 4,751,536 | G/GATA | 2.73 × 10−7 | Intronic | TOX3 | |
| 11 | 4,799,561 | CAG/C | 3.12 × 10−7 | Intronic | TOX3 | |
| 11 | 5,847,690 | TCGA/T | 1.04 × 10−6 | Intronic | CYLD | |
| TL | 1 | 183,023,926 | C/CT | 5.63 × 10−7 | Intergenic | TRPC6, PGR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Nong, Y.; Liu, Y.; Wang, Z.; Wang, J.; Li, Z. Genome-Wide Association Study of Body Size Traits in Luning Chickens Using Whole-Genome Sequencing. Animals 2025, 15, 972. https://doi.org/10.3390/ani15070972
Li Z, Nong Y, Liu Y, Wang Z, Wang J, Li Z. Genome-Wide Association Study of Body Size Traits in Luning Chickens Using Whole-Genome Sequencing. Animals. 2025; 15(7):972. https://doi.org/10.3390/ani15070972
Chicago/Turabian StyleLi, Zhiyi, Yi Nong, Yuan Liu, Zi Wang, Jiayan Wang, and Zhixiong Li. 2025. "Genome-Wide Association Study of Body Size Traits in Luning Chickens Using Whole-Genome Sequencing" Animals 15, no. 7: 972. https://doi.org/10.3390/ani15070972
APA StyleLi, Z., Nong, Y., Liu, Y., Wang, Z., Wang, J., & Li, Z. (2025). Genome-Wide Association Study of Body Size Traits in Luning Chickens Using Whole-Genome Sequencing. Animals, 15(7), 972. https://doi.org/10.3390/ani15070972