

Seasonal and Regional Dynamics of the Intestinal Microbiota in Schizothorax nukiangensis from the Nujiang River
 , , , ,
, , , ,  ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
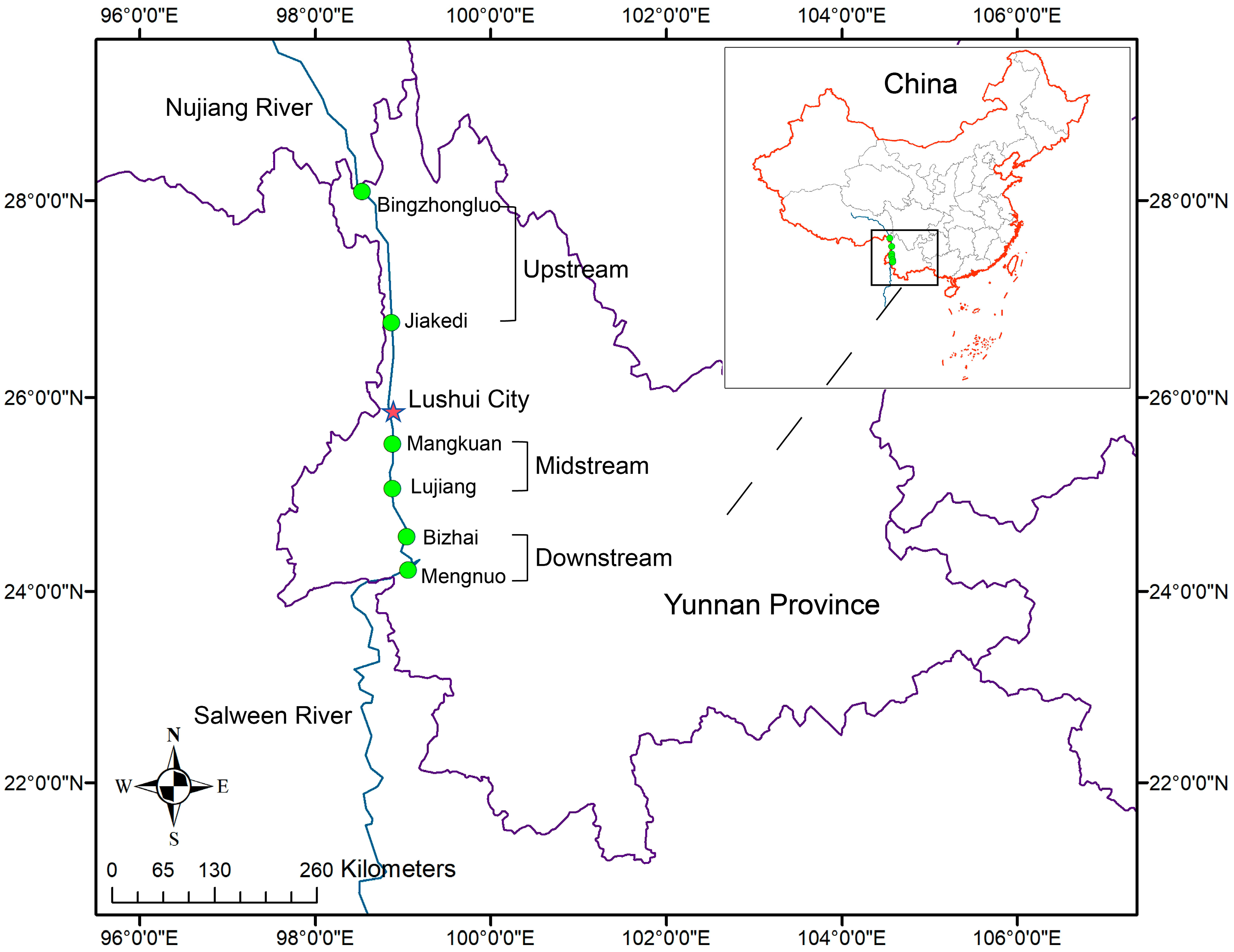
2.1. Sample Collection
2.2. DNA Extraction
2.3. Metagenome Sequencing, Data Preprocessing, and Assembly
2.4. Metagenomic and Statistical Analysis
3. Results
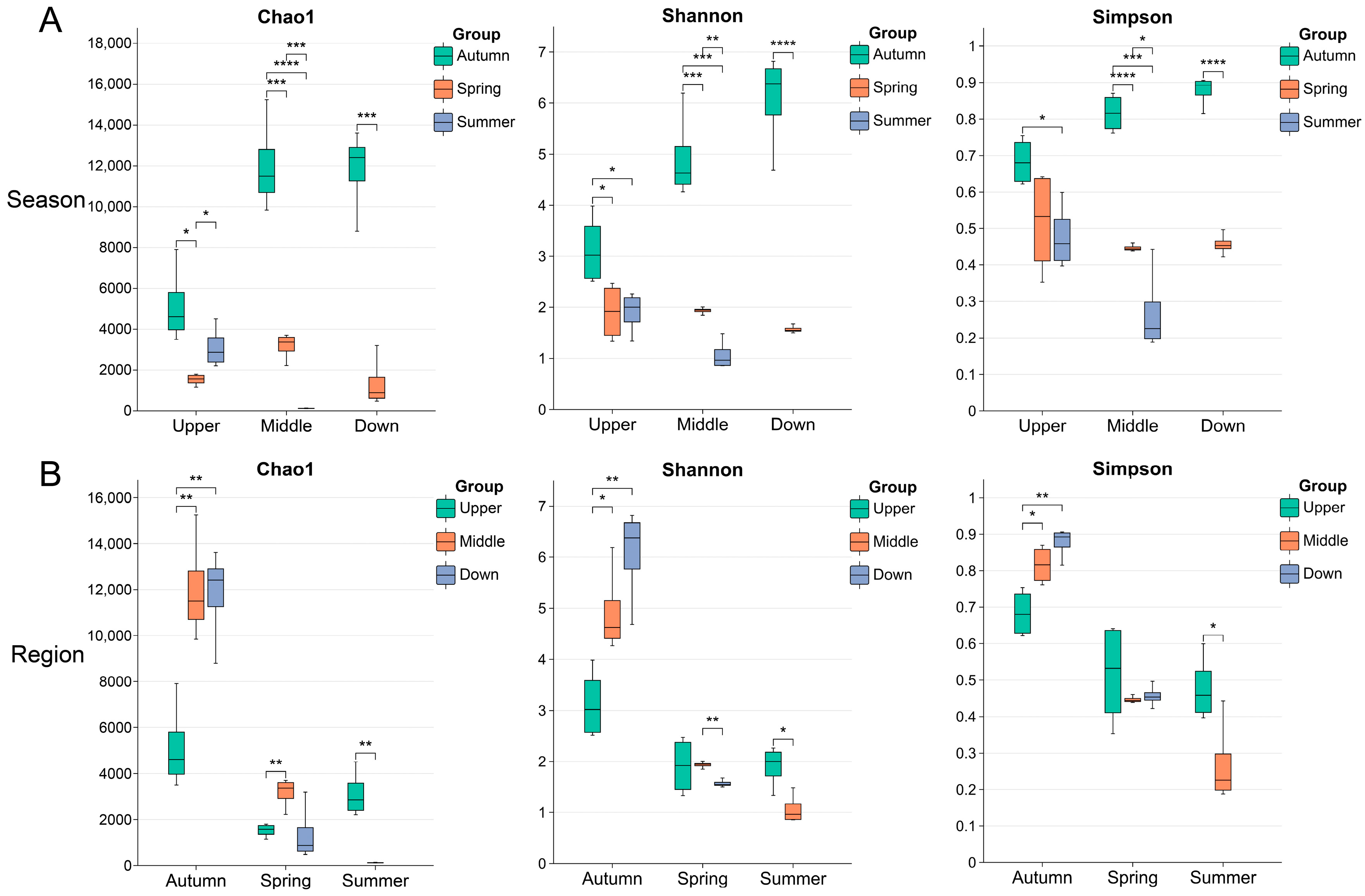
3.1. Overview of Metagenome Sequencing Results and Microbial Diversity
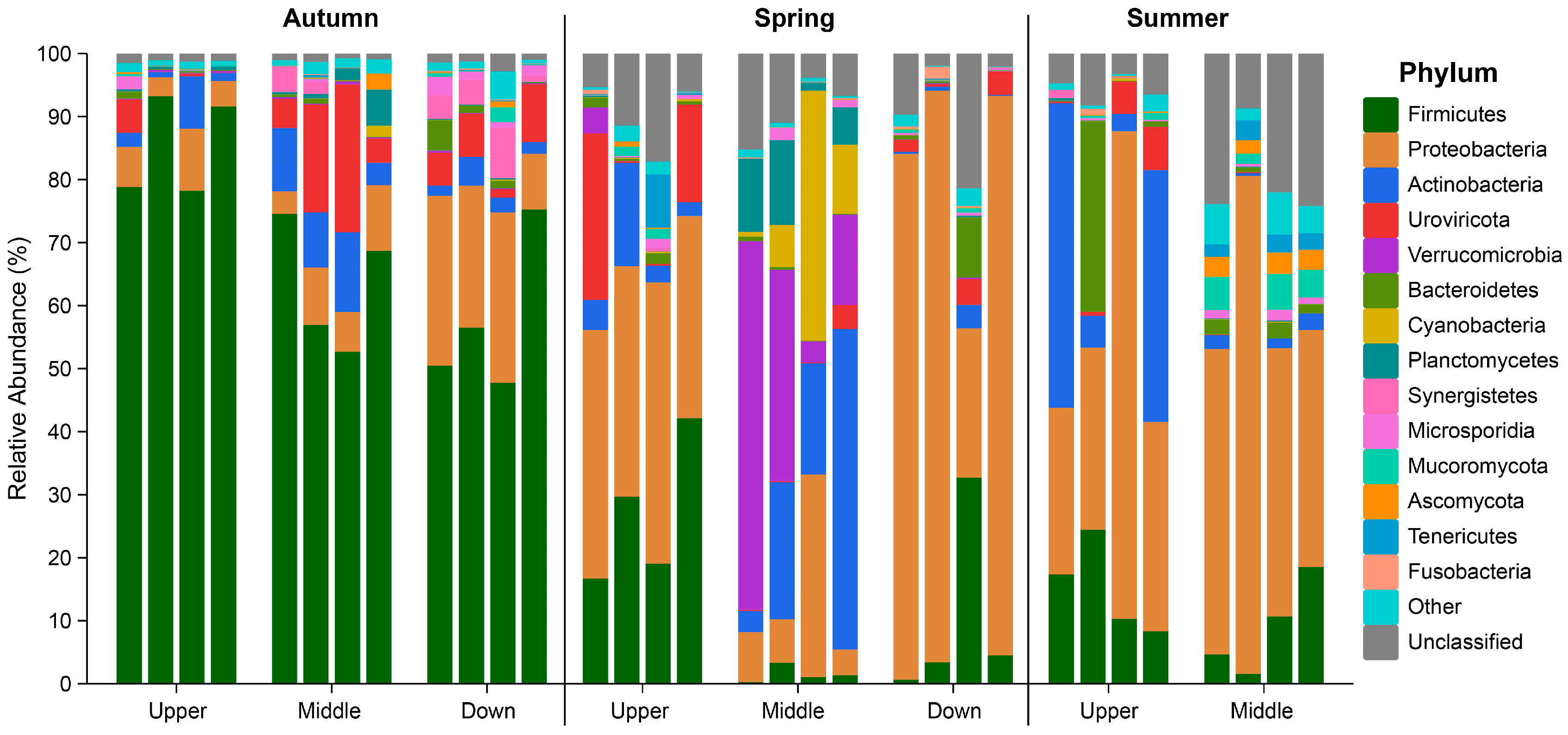
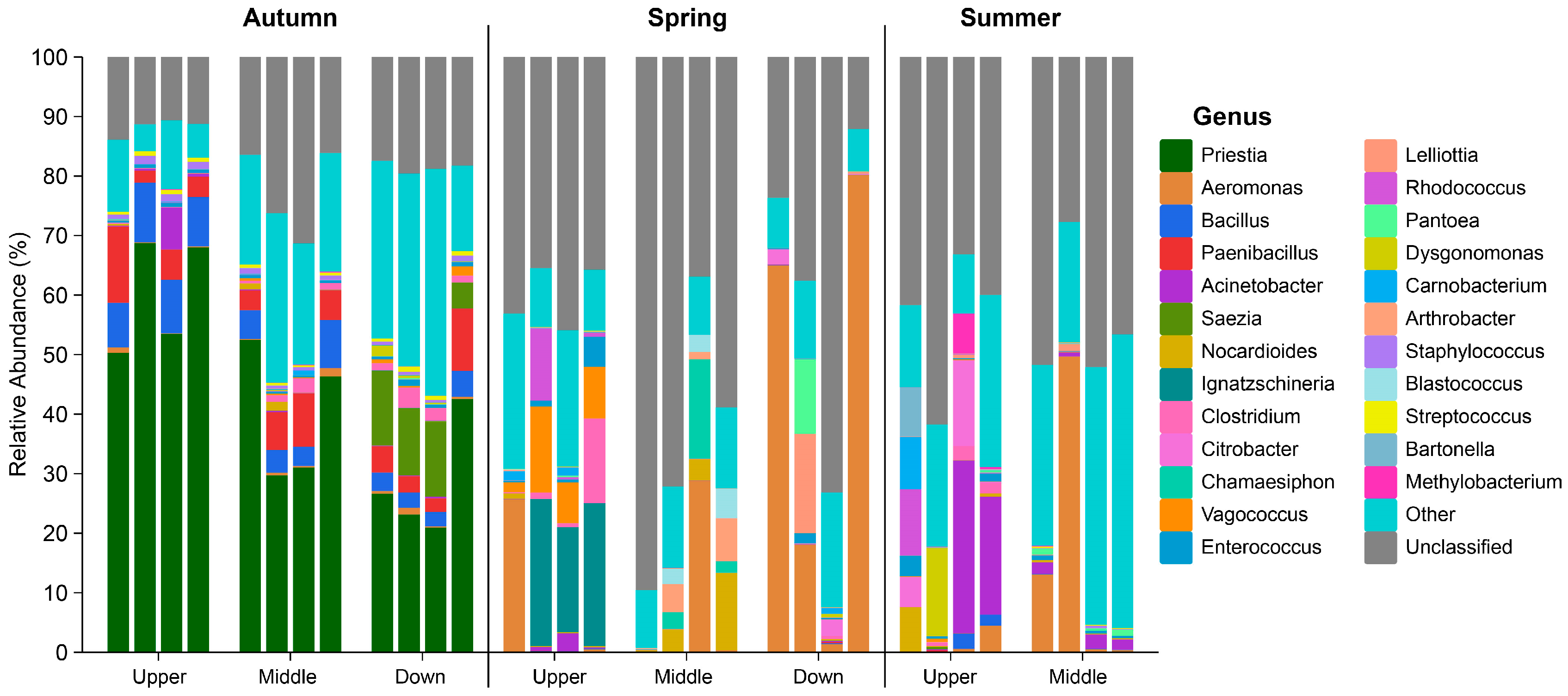
3.2. Composition of the Intestinal Microbiota in S. nukiangensis
3.3. Analysis of Biomarker Species in Intestinal Microbiota
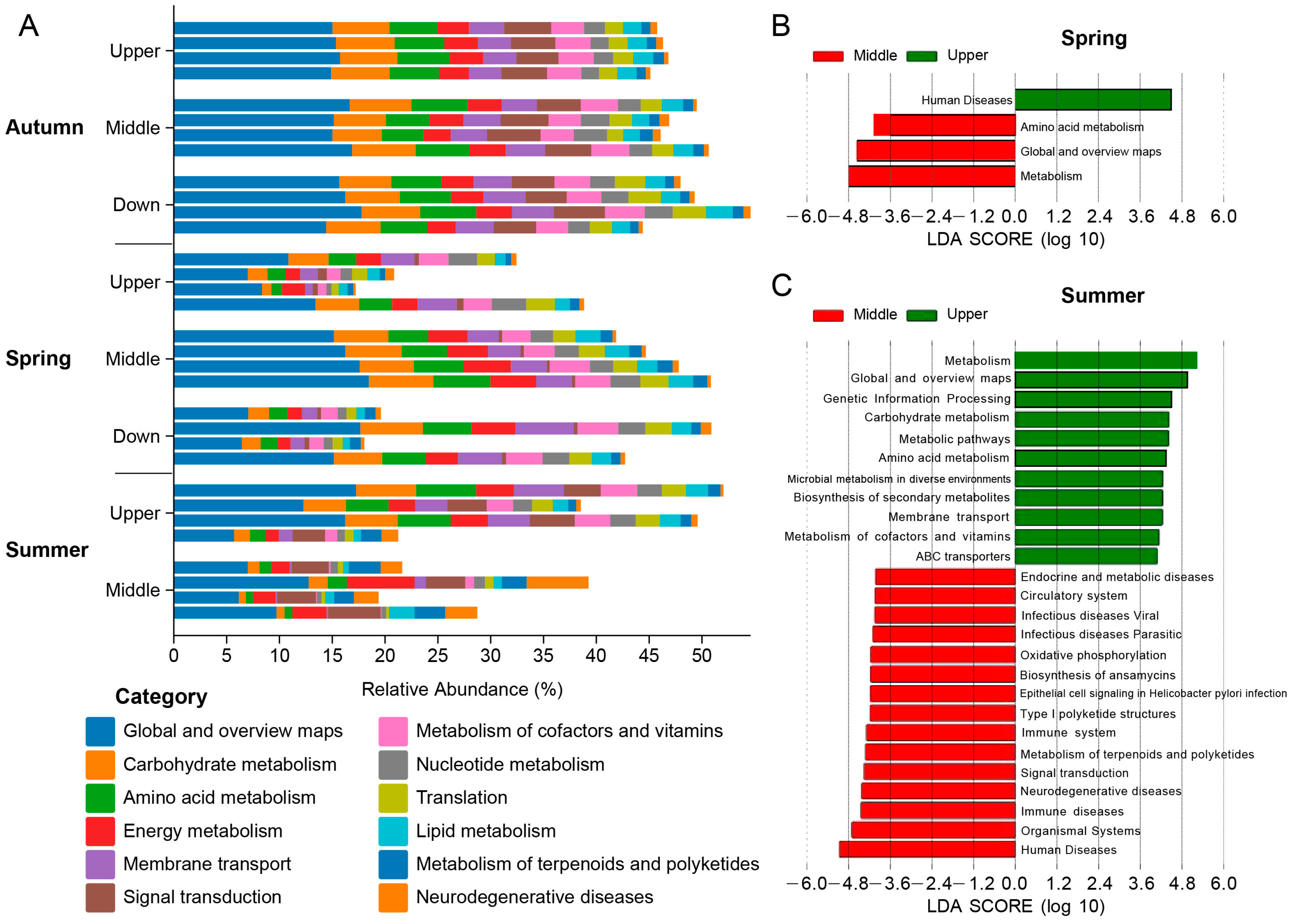
3.4. Functional Analysis of the Intestinal Microbiota in S. nukiangensis
4. Discussion
4.1. Seasonal Dynamics of the Intestinal Microbiota in S. nukiangensis
4.2. Regional Dynamics of the Intestinal Microbiota in S. nukiangensis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, N.; Wu, Q.; Shi, F.; Niu, J.; Zhang, T.; Degen, A.A.; Fang, Q.; Ding, L.; Shang, Z.; Zhang, Z.; et al. Seasonal dynamics of diet-gut microbiota interaction in adaptation of yaks to life at high altitude. NPJ Biofilms Microbiomes 2021, 7, 38. [Google Scholar] [CrossRef]
- Wong, L.L.; Mat Deris, Z.; Asaduzzaman, M.; Wang, M.; Liang, Y.; Sung, Y.Y.; Iehata, S. Gut microbiome variation based on 16S rRNA gene amplicon sequencing of the wild and domesticated broodstock populations of black tiger shrimp (Penaeus monodon) in the Indo-Pacific region. Ecol. Genet. Genom. 2023, 29, 100204. [Google Scholar] [CrossRef]
- Yan, Q.; Li, J.; Yu, Y.; Wang, J.; He, Z.; Van Nostrand, J.D.; Kempher, M.L.; Wu, L.; Wang, Y.; Liao, L.; et al. Environmental filtering decreases with fish development for the assembly of gut microbiota. Environ. Microbiol. 2016, 18, 4739–4754. [Google Scholar] [CrossRef] [PubMed]
- Vasemägi, A.; Visse, M.; Kisand, V. Effect of Environmental Factors and an Emerging Parasitic Disease on Gut Microbiome of Wild Salmonid Fish. mSphere 2017, 2, e00418-17. [Google Scholar] [CrossRef]
- Butt, R.L.; Volkoff, H. Gut Microbiota and Energy Homeostasis in Fish. Front. Endocrinol. 2019, 10, 9. [Google Scholar] [CrossRef]
- Ren, T.; Boutin, S.; Humphries, M.M.; Dantzer, B.; Gorrell, J.C.; Coltman, D.W.; McAdam, A.G.; Wu, M. Seasonal, spatial, and maternal effects on gut microbiome in wild red squirrels. Microbiome 2017, 5, 163. [Google Scholar] [CrossRef]
- Yang, H.; Wu, J.; Du, H.; Zhang, H.; Li, J.; Wei, Q. Quantifying the Colonization of Environmental Microbes in the Fish Gut: A Case Study of Wild Fish Populations in the Yangtze River. Front. Microbiol. 2021, 12, 828409. [Google Scholar] [CrossRef]
- Rothman, J.M.; Dierenfeld, E.S.; Hintz, H.F.; Pell, A.N. Nutritional quality of gorilla diets: Consequences of age, sex, and season. Oecologia 2008, 155, 111–122. [Google Scholar] [CrossRef]
- Greene, L.K.; Clayton, J.B.; Rothman, R.S.; Semel, B.P.; Semel, M.A.; Gillespie, T.R.; Wright, P.C.; Drea, C.M. Local habitat, not phylogenetic relatedness, predicts gut microbiota better within folivorous than frugivorous lemur lineages. Biol. Lett. 2019, 15, 20190028. [Google Scholar] [CrossRef]
- Li, S.; Qian, Z.; Yang, J.; Lin, Y.; Li, H.; Chen, L. Seasonal variation in structure and function of gut microbiota in Pomacea canaliculata. Ecol. Evol. 2022, 12, e9162. [Google Scholar] [CrossRef]
- Yang, K.; Ding, C.; Chen, X.; Ding, L.; Huang, M.; Chen, J.; Tao, J. Fish diversity and spatial distribution pattern in the Nujiang River Basin. Biodivers. Sci. 2022, 32, 21334. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, D. Asian river fishes in the Anthropocene: Threats and conservation challenges in an era of rapid environmental change. J. Fish Biol. 2011, 79, 1487–1524. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ma, B.; Zhang, C.; Tang, T.; Liu, S.; Duan, X.; Li, L.; Zhu, F.; Wang, N.; Chen, D. Distribution pattern and environmental impact factors of Schizothoracinae fishes in the rivers of Tibet: The case of Nujiang River and Yalu Zangbo River. Ecol. Environ. Sci. 2020, 29, 1792–1800. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, C.; Wu, L.; Luo, Z.; Sun, G.; Guo, Z.; Li, X.; Lin, F.; Chen, X. Fish diversity and threat factors in the Yunnan section of the Nujiang River. Biodivers. Sci. 2024, 32, 30–44. [Google Scholar] [CrossRef]
- Chen, W.; Du, K.; He, S. Genetic structure and historical demography of Schizothorax nukiangensis (Cyprinidae) in continuous habitat. Ecol. Evol. 2015, 5, 984–995. [Google Scholar] [CrossRef]
- Huang, F.J.; Liu, M.D.; Yu, L.X.; Liu, S.P. Length-weight relationships of five fish species from the Nujiang River (Salween basin), southwest China. J. Appl. Ichthyol. 2018, 34, 698–699. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, M.; Zhu, F.; Li, Z.; Liu, S.; Duan, X.; Chen, D.; Yang, R. Comparative Study of Three Species of Schizothoracine on Feeding and Digestive Organs in Upper Nujiang River. Chin. J. Zool. 2019, 54, 207–221. [Google Scholar] [CrossRef]
- Xu, W.; Zhu, F.; Wang, D.; Chen, D.; Duan, X.; Liu, M.; Li, D. Comparative Analysis of Metabolites between Different Altitude Schizothorax nukiangensis (Cyprinidae, Schizothoracine) on the Qinghai-Tibet Plateau in Nujiang River. Water 2023, 15, 284. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, Z.; Gao, N.; Qi, X.; Zeng, J.; Cui, K.; Lu, W.; Bai, S. Variations and Interseasonal Changes in the Gut Microbial Communities of Seven Wild Fish Species in a Natural Lake with Limited Water Exchange during the Closed Fishing Season. Microorganisms 2024, 12, 800. [Google Scholar] [CrossRef]
- Dulski, T.; Kozłowski, K.; Ciesielski, S. Habitat and seasonality shape the structure of tench (Tinca tinca L.) gut microbiome. Sci. Rep. 2020, 10, 4460. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Wang, S.; Wu, Y.; Sun, J.; Zhao, F. Seasonal dynamics of intestinal microbiota in juvenile Chinese mitten crab (Eriocheir sinensis) in the Yangtze Estuary. Front. Cell Infect. Microbiol. 2024, 14, 1436547. [Google Scholar] [CrossRef]
- Hovda, M.B.; Fontanillas, R.; McGurk, C.; Obach, A.; Rosnes, J.T. Seasonal variations in the intestinal microbiota of farmed Atlantic salmon (Salmo salar L.). Aquac. Res. 2012, 43, 154–159. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, W.; Sun, J.; Ma, W.; Chen, X.; Lian, Y. Temporal and Spatial Distribution of Hourly Precipitation in Rainy and Dry Seasons over Yunnan Province. Plateau Mt. Meteorol. Res. 2021, 41, 24–32. [Google Scholar] [CrossRef]
- Hao, J.; Zhang, M.; Zhang, Z.; Wang, X.; Liu, S.; Zhou, W. Effects of river water quality on bacterioplankton and pathogens communities. J. Shandong Univ. (Eng. Sci.) 2024, 54, 160–170. [Google Scholar] [CrossRef]
- Bahar-Tokman, H.; Demirci, M.; Keskin, F.E.; Cagatay, P.; Taner, Z.; Ozturk-Bakar, Y.; Ozyazar, M.; Kiraz, N.; Kocazeybek, B.S. Firmicutes/Bacteroidetes Ratio in the Gut Microbiota and IL-1β, IL-6, IL-8, TLR2, TLR4, TLR5 Gene Expressions in Type 2 Diabetes. Clin. Lab. 2022, 68, 1903. [Google Scholar] [CrossRef]
- Ravcheev, D.A.; Thiele, I. Comparative genomic analysis of the human gut microbiome reveals a broad distribution of metabolic pathways for the degradation of host-synthetized mucin glycans and utilization of mucin-derived monosaccharides. Front. Genet. 2017, 8, 111. [Google Scholar] [CrossRef]
- Mekuchi, M.; Asakura, T.; Sakata, K.; Yamaguchi, T.; Teruya, K.; Kikuchi, J. Intestinal microbiota composition is altered according to nutritional biorhythms in the leopard coral grouper (Plectropomus leopardus). PLoS ONE 2018, 13, e0197256. [Google Scholar] [CrossRef]
- Tran, N.T.; Xiong, F.; Hao, Y.-T.; Zhang, J.; Wu, S.-G.; Wang, G.-T. Starvation influences the microbiota assembly and expression of immunity-related genes in the intestine of grass carp (Ctenopharyngodon idellus). Aquaculture 2018, 489, 121–129. [Google Scholar] [CrossRef]
- Liu, X.; Shi, H.; He, Q.; Lin, F.; Wang, Q.; Xiao, S.; Dai, Y.; Zhang, Y.; Yang, H.; Zhao, H. Effect of starvation and refeeding on growth, gut microbiota and non-specific immunity in hybrid grouper (Epinephelus fuscoguttatus♀ × E. lanceolatus♂). Fish. Shellfish Immunol. 2020, 97, 182–193. [Google Scholar] [CrossRef]
- Li, Y.; Gajardo, K.; Jaramillo-Torres, A.; Kortner, T.M.; Krogdahl, Å. Consistent changes in the intestinal microbiota of Atlantic salmon fed insect meal diets. Anim. Microbiome 2022, 4, 8. [Google Scholar] [CrossRef]
- Hai, Q.; Wang, J.F.; Kang, W.G.; Lv, N.N.; Liu, Y.; Liu, Z.; Cheng, S.R. Analysis of Immune-related Gene Expression and Differences in Gut Microflora Between Two Sizes of Rainbow Trout (Oncorhynchus mykiss). J. Agric. Biotechnol. 2023, 31, 1043–1052. [Google Scholar] [CrossRef]
- Liu, X.; Dong, W.; Huang, H.; Liao, R.; Chen, Y.; Tan, B.; Lin, S. Effects of dietary high amylose on intestinal health of Micropterus salmoides. J. Fish. China 2023, 47, 109601–109610. [Google Scholar] [CrossRef]
- Ye, L.; Amberg, J.; Chapman, D.; Gaikowski, M.; Liu, W.T. Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous. Am. Fish. ISME J. 2014, 8, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.H.; Ahmad, M.; Hardiman, E.M.; Rahmanpour, R. Pathways for degradation of lignin in bacteria and fungi. Nat. Prod. Rep. 2011, 28, 1883–1896. [Google Scholar] [CrossRef]
- Sharma, A.; Chanu, T.I.; Nayak, S.K.; Jahageerdar, S.; Krishna, G. Pathogenesis of Aeromonas caviae in Clarias magur. Microb. Pathog. 2022, 169, 105662. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, N.; Li, L.; Qiang, Z.; Wei, J.; Nie, Z. Analysis of the diet of 5 indigenous fishes in Kizil River, Xinjiang. J. Fish. Sci. China 2022, 29, 1461–1474. [Google Scholar] [CrossRef]
- Jiang, M.; Xu, M.; Ying, C.; Yin, D.; Dai, P.; Yang, Y.; Ye, K.; Liu, K. The intestinal microbiota of lake anchovy varies according to sex, body size, and local habitat in Taihu Lake, China. Microbiologyopen 2020, 9, e00955. [Google Scholar] [CrossRef]
- Kim, P.S.; Shin, N.R.; Lee, J.B.; Kim, M.S.; Whon, T.W.; Hyun, D.W.; Yun, J.H.; Jung, M.J.; Kim, J.Y.; Bae, J.W. Host habitat is the major determinant of the gut microbiome of fish. Microbiome 2021, 9, 166. [Google Scholar] [CrossRef]
- Medina-Pascual, M.J.; Monzón, S.; Villalón, P.; Cuesta, I.; González-Romo, F.; Valdezate, S. Saezia sanguinis gen. nov., sp. nov., a Betaproteobacteria member of order Burkholderiales, isolated from human blood. Int. J. Syst. Evol. Microbiol. 2020, 70, 2016–2025. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Dutkiewicz, J.; Mackiewicz, B.; Lemieszek, M.K.; Golec, M.; Milanowski, J. Pantoea agglomerans: A mysterious bacterium of evil and good. Part III. Deleterious effects: Infections of humans, animals and plants. Ann. Agric. Environ. Med. 2016, 23, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tang, Y.; Wang, D.; Lin, N.; Zhou, J. Identification and characterization of a New Enterobacter onion bulb decay caused by Lelliottia amnigena in China. Appl. Microbiol. 2016, 2, 1–7. [Google Scholar] [CrossRef]
- Osei, R.; Yang, C.; Cui, L.; Ma, T.; Li, Z.; Boamah, S. Isolation, identification, and pathogenicity of Lelliottia amnigena causing soft rot of potato tuber in China. Microb. Pathog. 2022, 164, 105441. [Google Scholar] [CrossRef]
- Menke, S.; Wasimuddin; Meier, M.; Melzheimer, J.; Mfune, J.K.; Heinrich, S.; Thalwitzer, S.; Wachter, B.; Sommer, S. Oligotyping reveals differences between gut microbiomes of free-ranging sympatric Namibian carnivores (Acinonyx jubatus, Canis mesomelas) on a bacterial species-like level. Front. Microbiol. 2014, 5, 526. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Relative Abundance (%) | |||
|---|---|---|---|---|
| Autumn | Spring | Summer | Average | |
| Firmicutes | 68.7 ± 15.7 A | 12.9 ± 14.8 B | 12.0 ± 7.6 B | 33.6 |
| Proteobacteria | 11.5 ± 8.9 B | 40.9 ± 31.1 A | 46.7 ± 20.7 A | 31.3 |
| Actinobacteria | 4.8 ± 4.0 | 10.4 ± 14.8 | 12.9 ± 19.5 | 8.9 |
| Uroviricota | 6.4 ± 7.2 | 4.7 ± 8.1 | 1.6 ± 2.7 | 4.6 |
| Verrucomicrobia | 0.2 ± 0.1 | 9.5 ± 18.3 | 0.0 ± 0.0 | 3.7 |
| Bacteroidetes | 0.2 ± 0.5 | 4.9 ± 11.5 | 0.1 ± 0.1 | 2.0 |
| Cyanobacteria | 0.9 ± 1.6 | 2.8 ± 4.9 | 0.1 ± 0.1 | 2.0 |
| Planctomycetes | 0.9 ± 1.3 | 1.3 ± 2.6 | 4.8 ± 10.3 | 1.4 |
| Sum | 93.7 ± 5.0 | 87.4 ± 8.9 | 78.2 ± 16.7 | 87.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, F.; Ma, J.; Xue, M.; Xu, W.; Liu, W.; Zhou, Y.; Liu, M.; Fan, Y. Seasonal and Regional Dynamics of the Intestinal Microbiota in Schizothorax nukiangensis from the Nujiang River. Animals 2025, 15, 961. https://doi.org/10.3390/ani15070961
Zhu F, Ma J, Xue M, Xu W, Liu W, Zhou Y, Liu M, Fan Y. Seasonal and Regional Dynamics of the Intestinal Microbiota in Schizothorax nukiangensis from the Nujiang River. Animals. 2025; 15(7):961. https://doi.org/10.3390/ani15070961
Chicago/Turabian StyleZhu, Fengyue, Jie Ma, Mingyang Xue, Weitong Xu, Wenzhi Liu, Yong Zhou, Mingdian Liu, and Yuding Fan. 2025. "Seasonal and Regional Dynamics of the Intestinal Microbiota in Schizothorax nukiangensis from the Nujiang River" Animals 15, no. 7: 961. https://doi.org/10.3390/ani15070961
APA StyleZhu, F., Ma, J., Xue, M., Xu, W., Liu, W., Zhou, Y., Liu, M., & Fan, Y. (2025). Seasonal and Regional Dynamics of the Intestinal Microbiota in Schizothorax nukiangensis from the Nujiang River. Animals, 15(7), 961. https://doi.org/10.3390/ani15070961