

Comparison of Transcriptome Differences in Scales of Two Closely Related Snake Species (Lycodon rufozonatus and Lycodon rosozonatus)

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
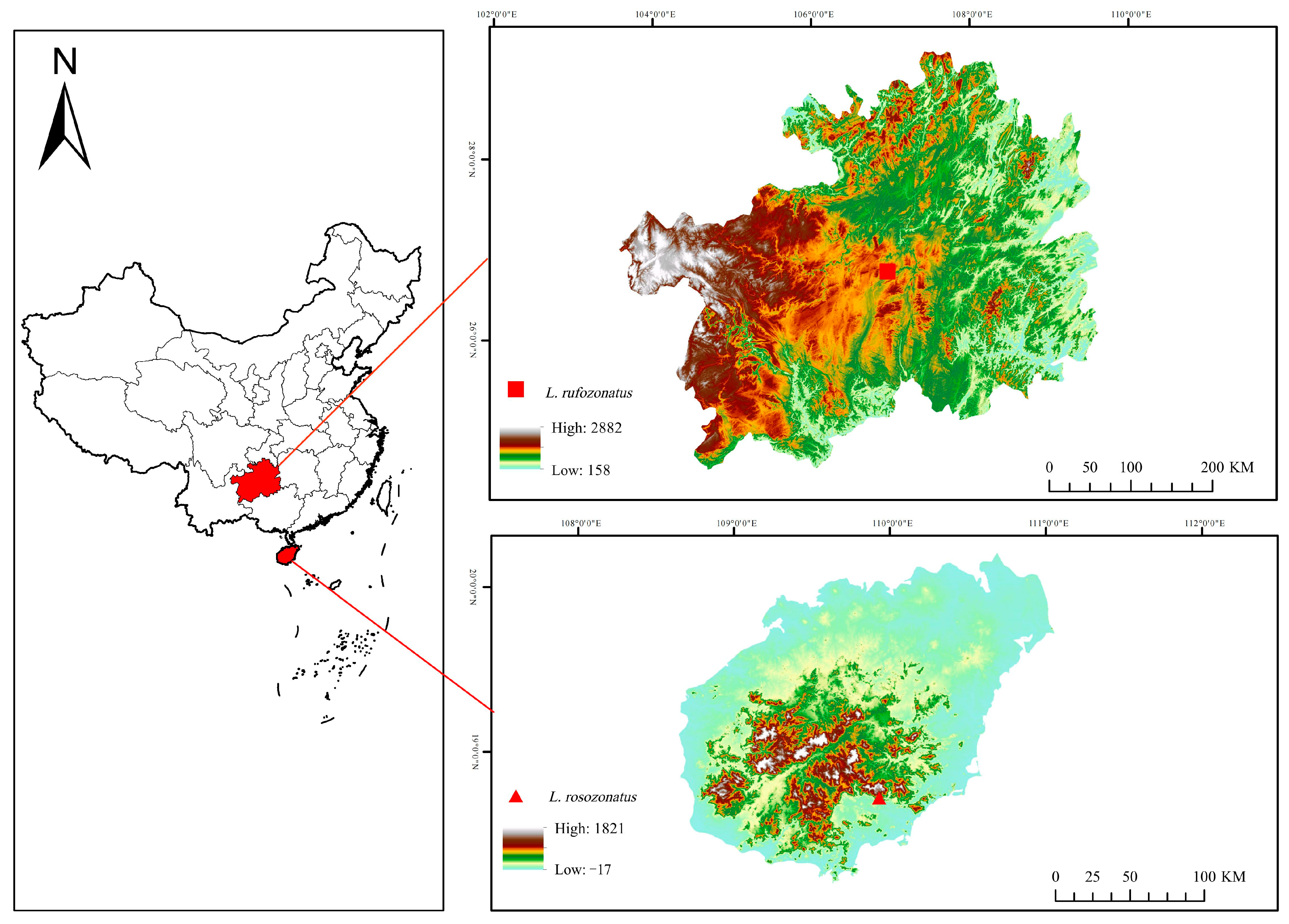
2.1. Animal Collection
2.2. Whole-Genome Resequencing and RNA Extraction
2.3. Transcriptome Assembly and Functional Annotation
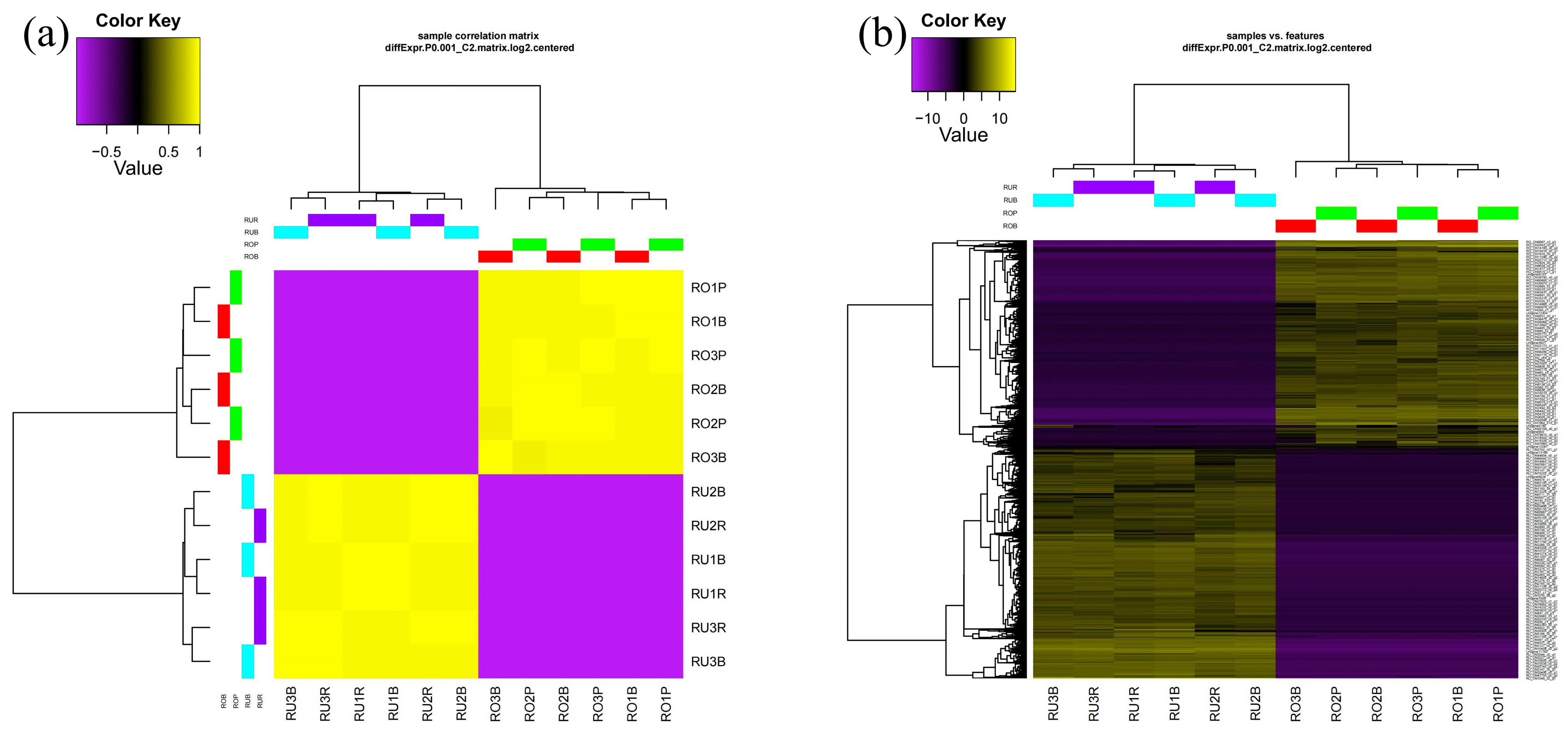
2.4. Correlation Analysis
2.5. Differential Expression Analysis
2.6. Single Amino Acid Polymorphism (SAAP) Analysis of Scale Color Differences
3. Results
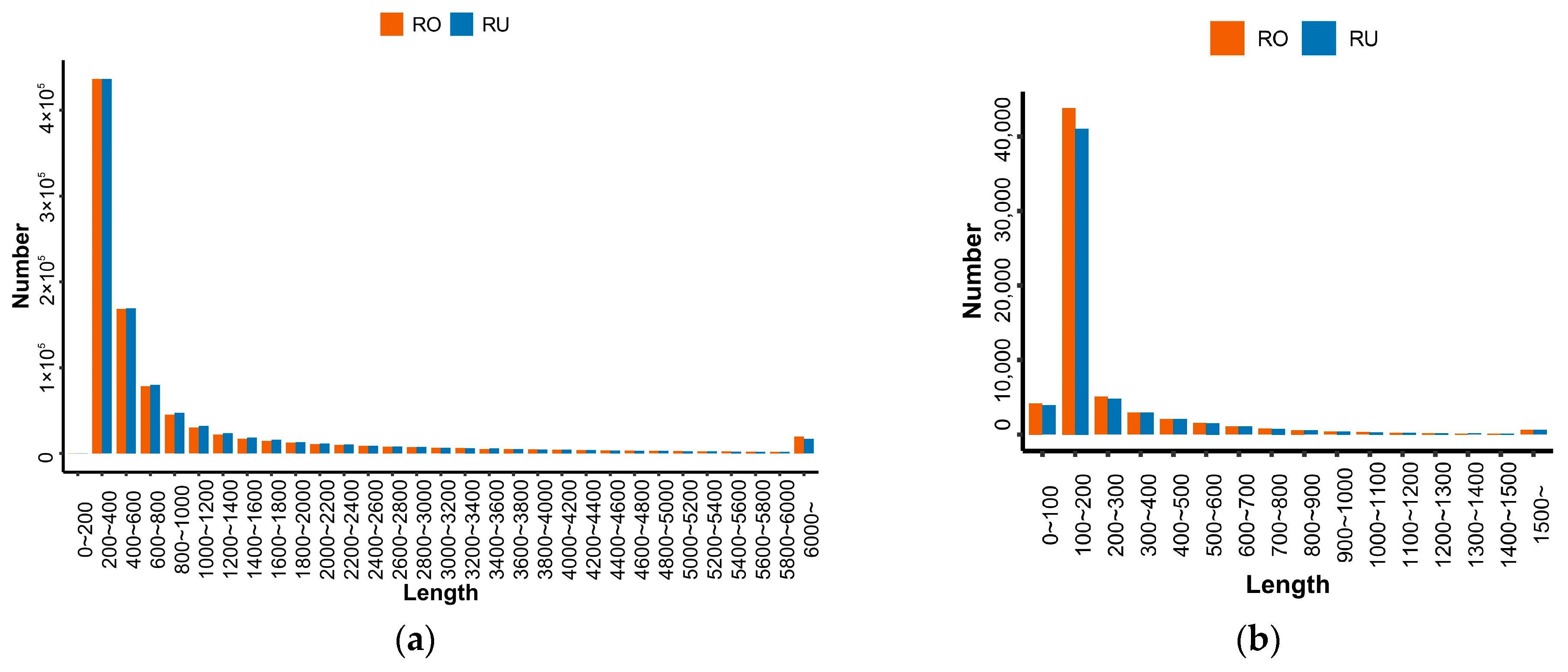
3.1. Whole-Genome Transcriptome Sequencing and Assembly
3.2. Annotation Analysis
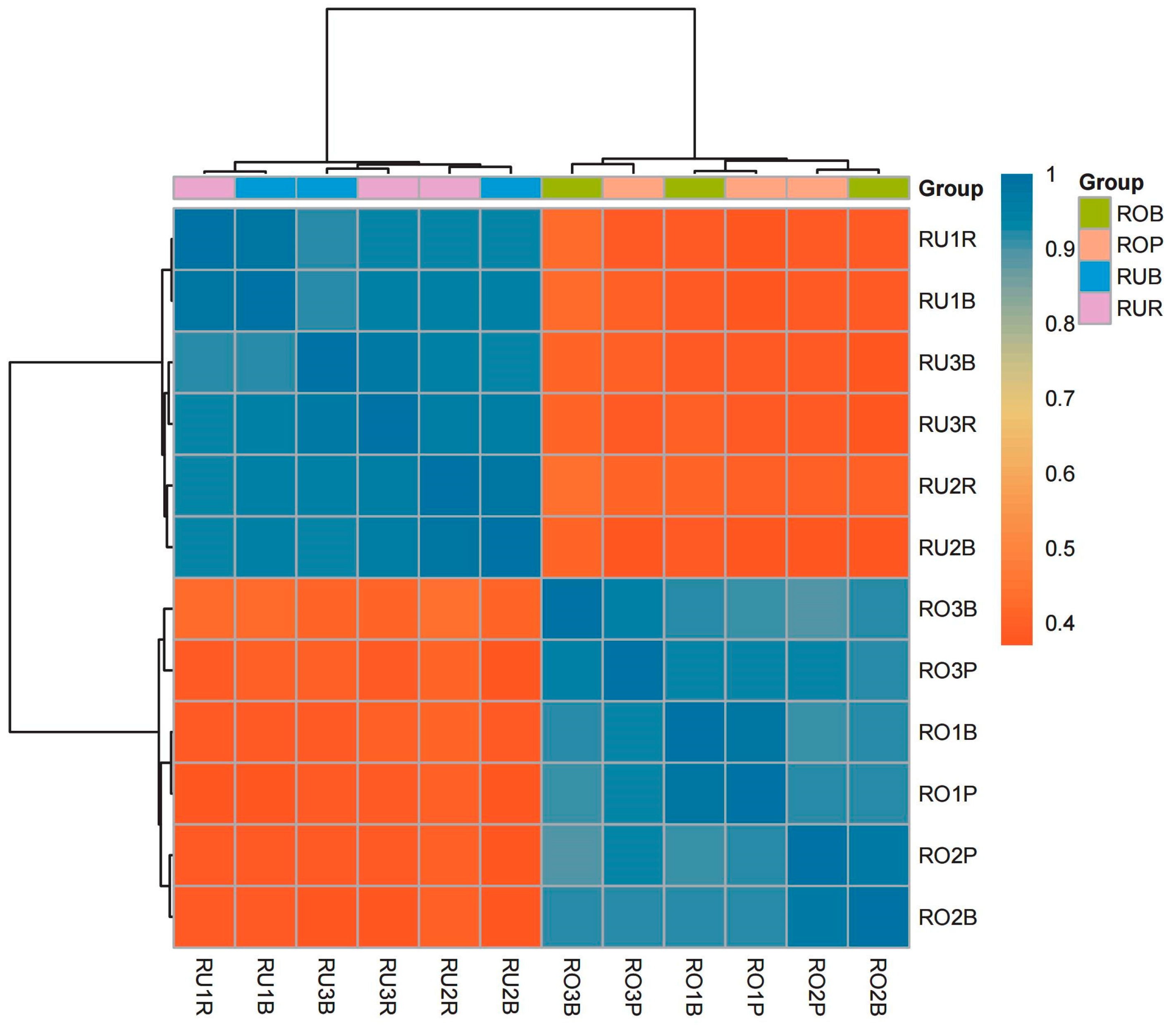
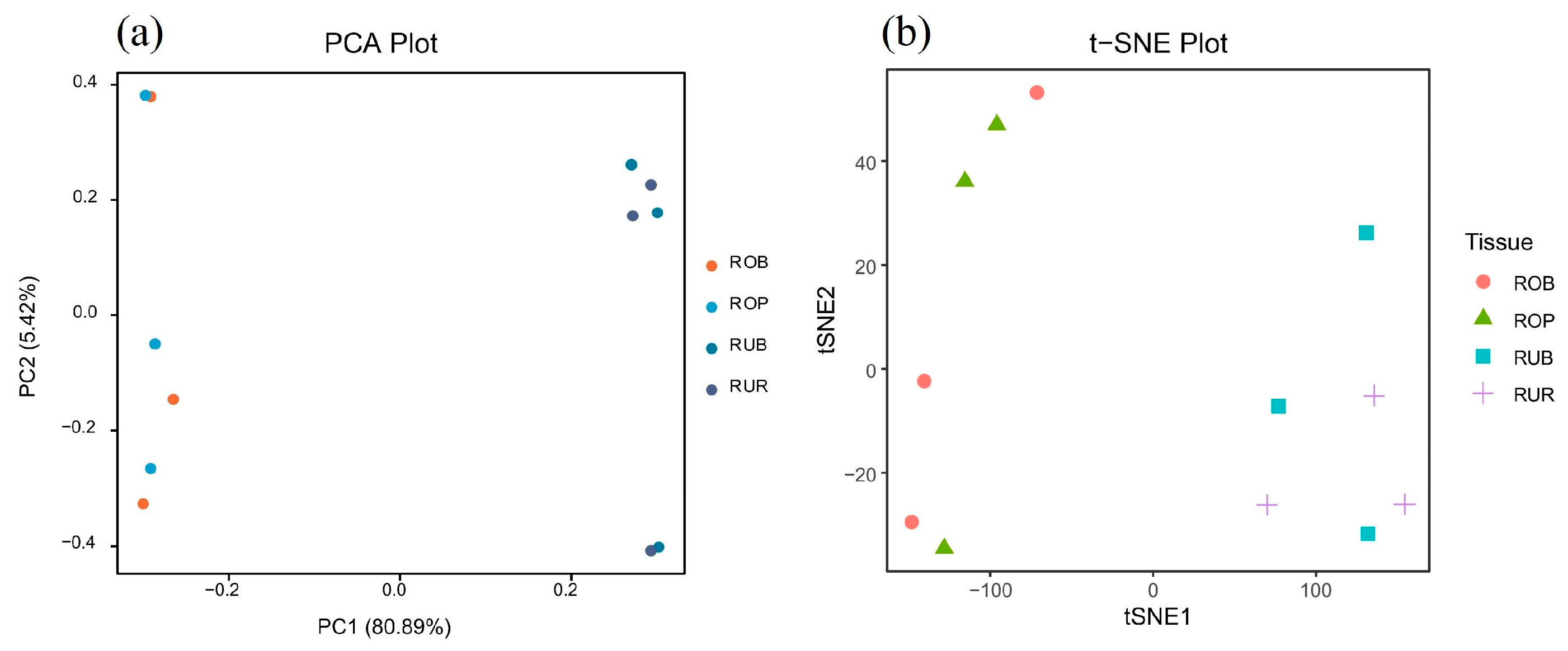
3.3. Correlation Analysis of Scales of Different Colors
3.4. DEGs in Different Transcriptomes
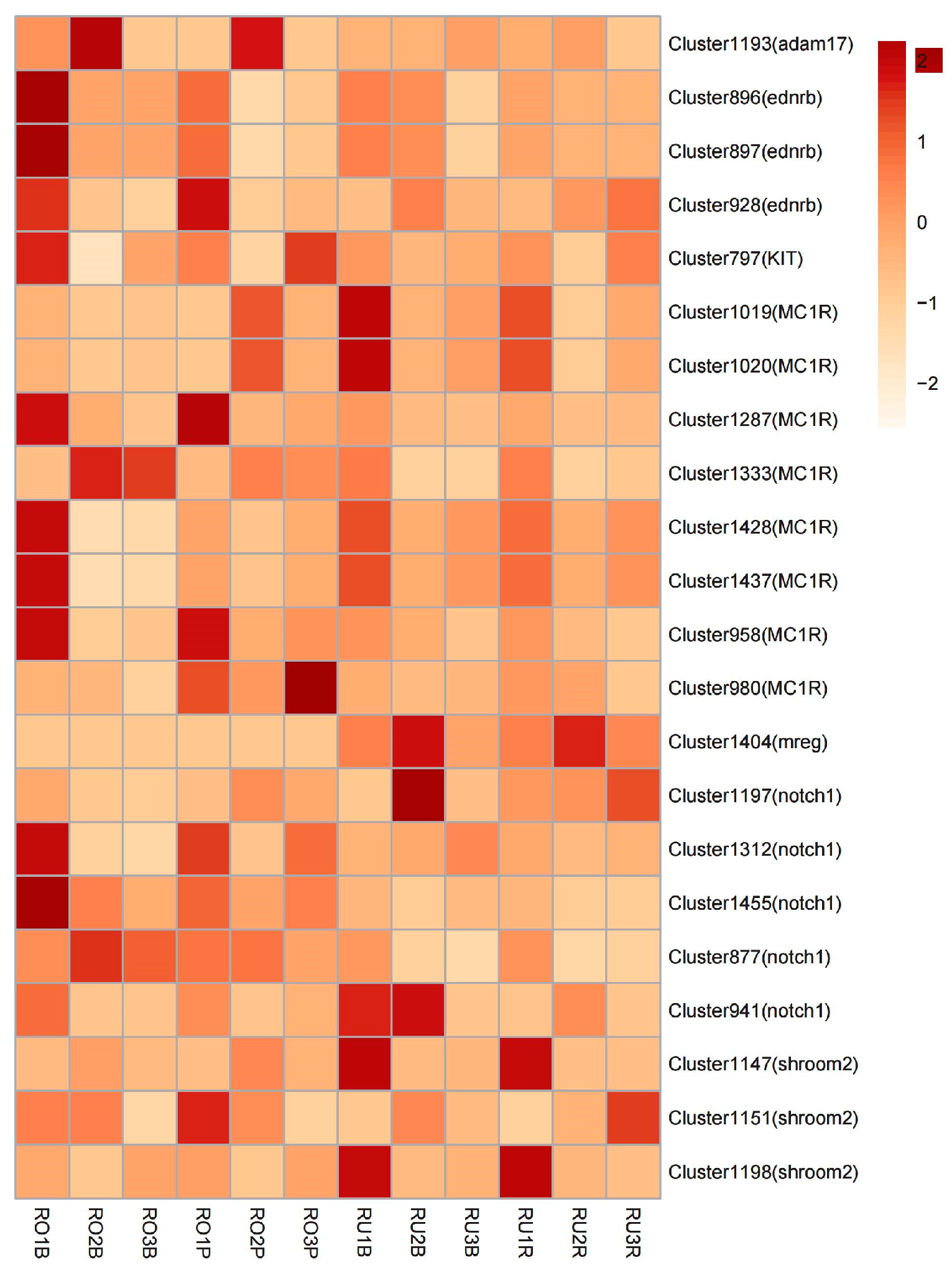
3.5. SAAP Analysis of Differentially Expressed Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hubbard, J.K.; Uy, J.A.C.; Hauber, M.E.; Hoekstra, H.E.; Safran, R.J. Vertebrate pigmentation: From underlying genes to adaptive function. Trends Genet. 2010, 26, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Protas, M.E.; Patel, N.H. Evolution of coloration patterns. Annu. Rev. Cell Dev. Biol. 2008, 24, 425–446. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, G.M.; Kelley, J.L.; Morrell, L.J. Colour change and assortment in the western rainbowfish. Anim. Behav. 2010, 79, 1025–1030. [Google Scholar] [CrossRef]
- Kelsh, R.N.; Harris, M.L.; Colanesi, S.; Erickson, C.A. Stripes and belly-spots—A review of pigment cell morphogenesis in vertebrates. Semin. Cell Dev. Biol. 2009, 20, 90–104. [Google Scholar] [CrossRef]
- Mills, M.G.; Patterson, L.B. Not just black and white: Pigment pattern development and evolution in vertebrates. Semin. Cell Dev. Biol. 2009, 20, 72–81. [Google Scholar] [CrossRef]
- Cuthill, I.C.; Allen, W.L.; Arbuckle, K.; Caspers, B.; Chaplin, G.; Hauber, M.E.; Hill, G.E.; Jablonski, N.G.; Jiggins, C.D.; Kelber, A.; et al. The biology of color. Science 2017, 357, eaan0221. [Google Scholar] [CrossRef]
- Bagnara, J.T.; Matsumoto, J.; Ferris, W.; Frost, S.K.; Turner, W.A.; Tchen, T.T.; Taylor, J.D. Common Origin of Pigment Cells. Science 1979, 203, 410–415. [Google Scholar] [CrossRef]
- Saenko, S.V.; Teyssier, J.; van der Marel, D.; Milinkovitch, M.C. Precise colocalization of interacting structural and pigmentary elements generates extensive color pattern variation in Phelsumalizards. BMC Biol. 2013, 11, 105. [Google Scholar] [CrossRef]
- Ducrest, A.-L.; Ursenbacher, S.; Golay, P.; Monney, J.-C.; Mebert, K.; Roulin, A.; Dubey, S. Pro-opiomelanocortin gene and melanin-based colour polymorphism in a reptile. Biol. J. Linn. Soc. 2013, 111, 160–168. [Google Scholar] [CrossRef]
- Gebreselassie, G.; Liang, B.; Berihulay, H.; Islam, R.; Abied, A.; Jiang, L.; Zhao, Z.; Ma, Y. Genomic mapping identifies two genetic variants in the MC1R gene for coat colour variation in Chinese Tan sheep. PLoS ONE 2020, 15, e0235426. [Google Scholar] [CrossRef]
- Norris, B.J.; Whan, V.A. A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 2008, 18, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Elfring, A.; Sabin, C.E.; Iouchmanov, A.L.; Roffey, H.L.; Samudra, S.P.; Alcala, A.J.; Osman, R.S.; Lauderdale, J.D.; Hendry, A.P.; Menke, D.B.; et al. Piebaldism and chromatophore development in reptiles are linked to the tfec gene. Curr. Biol. 2023, 33, 755–763.e3. [Google Scholar] [CrossRef] [PubMed]
- Ghiradella, H. Structure and Development of Iridescent Lepidopteran Scales: The Papilionidae as a Showcase Family. Ann. Entomol. Soc. Am. 1985, 78, 252–264. [Google Scholar]
- Kinoshita, S.; Yoshioka, S. Structural colors in nature: The role of regularity and irregularity in the structure. Chemphyschem 2005, 6, 1442–1459. [Google Scholar] [CrossRef]
- Tang, C.-Y.; Zhang, X.; Xu, X.; Sun, S.; Peng, C.; Song, M.-H.; Yan, C.; Sun, H.; Liu, M.; Xie, L.; et al. Genetic mapping and molecular mechanism behind color variation in the Asian vine snake. Genome Biol. 2023, 24, 46. [Google Scholar] [CrossRef]
- Boback, S.M.; Siefferman, L.M. Variation in Color and Color Change in Island and Mainland Boas (Boa constrictor). J. Herpetol. 2010, 44, 506–515. [Google Scholar] [CrossRef]
- McLean, C.A.; Stuart-Fox, D.; Moussalli, A. Environment, but not genetic divergence, influences geographic variation in colour morph frequencies in a lizard. BMC Evol. Biol. 2015, 15, 156. [Google Scholar] [CrossRef]
- Corl, A.; Davis, A.R.; Kuchta, S.R.; Sinervo, B. Selective loss of polymorphic mating types is associated with rapid phenotypic evolution during morphic speciation. Proc. Natl. Acad. Sci. USA 2010, 107, 4254–4259. [Google Scholar] [CrossRef]
- Hugall, A.F.; Stuart-Fox, D. Accelerated speciation in colour-polymorphic birds. Nature 2012, 485, 631–634. [Google Scholar] [CrossRef]
- McLean, C.A.; Stuart-Fox, D. Geographic variation in animal colour polymorphisms and its role in speciation. Biol. Rev. Camb. Philos. Soc. 2014, 89, 860–873. [Google Scholar] [CrossRef]
- Geneva, A.J.; Hilton, J.; Noll, S.; Glor, R.E. Multilocus phylogenetic analyses of Hispaniolan and Bahamian trunk anoles (distichus species group). Mol. Phylogenetics Evol. 2015, 87, 105–117. [Google Scholar] [CrossRef] [PubMed]
- MacGuigan, D.J.; Geneva, A.J.; Glor, R.E. A genomic assessment of species boundaries and hybridization in a group of highly polymorphic anoles (distichus species complex). Ecol. Evol. 2017, 7, 3657–3671. [Google Scholar] [CrossRef] [PubMed]
- Myers, T.C.; de Mello, P.L.H.; Glor, R.E. A morphometric assessment of species boundaries in a widespread anole lizard (Squamata: Dactyloidae). Biol. J. Linn. Soc. 2020, 130, 813–825. [Google Scholar] [CrossRef]
- Srikanthan, A.N.; Adhikari, O.D.; Mallik, A.K.; Campbell, P.D.; Bhatt, B.B.; Shanker, K.; Ganesh, S.R. Taxonomic revaluation of the Ahaetulla prasina (H. Boie in F. Boie, 1827) complex from Northeast India: Resurrection and redescription of Ahaetulla flavescens (Wall, 1910) (Reptilia: Serpentes: Colubridae). Eur. J. Taxon. 2022, 839, 120–148. [Google Scholar] [CrossRef]
- Murakami, A.; Hasegawa, M.; Kuriyama, T. Developmental mechanisms of longitudinal stripes in the Japanese four-lined snake. J. Morphol. 2018, 279, 27–36. [Google Scholar] [CrossRef]
- I de Lanuza, G.P.; Carazo, P.; Font, E. Colours of quality: Structural (but not pigment) coloration informs about male quality in a polychromatic lizard. Anim. Behav. 2014, 90, 73–81. [Google Scholar] [CrossRef]
- Rosenblum, E.B.; Hoekstra, H.E.; Nachman, M.W. Adaptive reptile color variation and the evolution of the MCIR gene. Evol. Int. J. Org. Evol. 2004, 58, 1794–1808. [Google Scholar] [CrossRef]
- McLean, C.A.; Lutz, A.; Rankin, K.J.; Stuart-Fox, D.; Moussalli, A. Revealing the Biochemical and Genetic Basis of Color Variation in a Polymorphic Lizard. Mol. Biol. Evol. 2017, 34, 1924–1935. [Google Scholar] [CrossRef]
- Kuriyama, T.; Misawa, H.; Miyaji, K.; Sugimoto, M.; Hasegawa, M. Pigment cell mechanisms underlying dorsal color-pattern polymorphism in the japanese four-lined snake. J. Morphol. 2013, 274, 1353–1364. [Google Scholar] [CrossRef]
- Lu, B.; Qiu, X.; Yang, W.; Yao, Z.; Ma, X.; Deng, S.; Zhang, Q.; Fu, J.; Qi, Y. Genetic Basis and Evolutionary Forces of Sexually Dimorphic Color Variation in a Toad-Headed Agamid Lizard. Mol. Biol. Evol. 2024, 41, msae054. [Google Scholar] [CrossRef]
- Dao, U.M.; Lederer, I.; Tabor, R.L.; Shahid, B.; Graves, C.W.; Seidel, H.S. Stripes and loss of color in ball pythons (Python regius) are associated with variants affecting endothelin signaling. G3 Genes|Genomes|Genet. 2023, 13, jkad063. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.L.; Baddeley, R.; Scott-Samuel, N.E.; Cuthill, I.C. The evolution and function of pattern diversity in snakes. Behav. Ecol. 2013, 24, 1237–1250. [Google Scholar] [CrossRef]
- Olsson, M.; Stuart-Fox, D.; Ballen, C. Genetics and evolution of colour patterns in reptiles. Semin. Cell Dev. Biol. 2013, 24, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Pizzigalli, C.; Banfi, F.; Ficetola, G.F.; Falaschi, M.; Mangiacotti, M.; Sacchi, R.; Zuffi, M.A.L.; Scali, S. Eco-geographical determinants of the evolution of ornamentation in vipers. Biol. J. Linn. Soc. 2020, 130, 345–358. [Google Scholar] [CrossRef]
- McLean, C.A.; Moussalli, A.; Sass, S.; Stuart-Fox, D. Taxonomic Assessment of the Ctenophorus decresii Complex (Reptilia: Agamidae) Reveals a New Species of Dragon Lizard from Western New South Wales. Rec. Aust. Mus. 2013, 65, 51–63. [Google Scholar] [CrossRef]
- McLean, C.A.; Stuart-Fox, D.; Moussalli, A. Phylogeographic structure, demographic history and morph composition in a colour polymorphic lizard. J. Evol. Biol. 2014, 27, 2123–2137. [Google Scholar] [CrossRef]
- Wang, K.; Ren, J.; Chen, H.; Lyu, Z.; Guo, X.; Jiang, K.; Chen, J.; Li, J.; Guo, P.; Wang, Y.; et al. The updated checklists of amphibians and reptiles of China. Biodivers. Sci. 2020, 28, 189–218. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L.; et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 2020, 9, giaa094. [Google Scholar] [CrossRef] [PubMed]
- Gremme, G.; Brendel, V.; Sparks, M.E.; Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 2005, 47, 965–978. [Google Scholar] [CrossRef]
- Sangrador-Vegas, A.; Mitchell, A.L.; Chang, H.-Y.; Yong, S.-Y.; Finn, R.D. GO annotation in InterPro: Why stability does not indicate accuracy in a sea of changing annotations. Database 2016, 2016, baw027. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Stuckert, A.M.M.; Moore, E.; Coyle, K.P.; Davison, I.; MacManes, M.D.; Roberts, R.; Summers, K. Variation in pigmentation gene expression is associated with distinct aposematic color morphs in the poison frog Dendrobates auratus. BMC Evol. Biol. 2019, 19, 85. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Altenhoff, A.M.; Levy, J.; Zarowiecki, M.; Tomiczek, B.; Vesztrocy, A.W.; Dalquen, D.A.; Müller, S.; Telford, M.J.; Glover, N.M.; Dylus, D.; et al. OMA standalone: Orthology inference among public and custom genomes and transcriptomes. Genome Res. 2019, 29, 1152–1163. [Google Scholar] [CrossRef]
- Kuriyama, T.; Murakami, A.; Brandley, M.C.; Hasegawa, M. Blue, Black, and Stripes: Evolution and Development of Color Production and Pattern Formation in Lizards and Snakes. Front. Ecol. Evol. 2020, 8, 232. [Google Scholar] [CrossRef]
- Pfennig, D.W.; Harcombe, W.R.; Pfennig, K.S. Frequency-dependent Batesian mimicry. Nature 2001, 410, 323. [Google Scholar] [CrossRef]
- Kodandaramaiah, U.; Palathingal, S.; Kurup, G.B.; Murali, G. What Makes Motion Dazzle Markings Effective Against Predation? Behav. Ecol. 2019, 31, 43–53. [Google Scholar] [CrossRef]
- Murali, G.; Merilaita, S.; Kodandaramaiah, U. Grab my tail: Evolution of dazzle stripes and colourful tails in lizards. J. Evol. Biol. 2018, 31, 1675–1688. [Google Scholar] [CrossRef] [PubMed]
- Bagnara, J.T.; Hadley, M.E. Chromatophores and Color Change: The Comparative Physiology of Animal Pigmentation; Prentice-Hall: Englewood Cliffs, NJ, USA, 1973. [Google Scholar]
- Kuriyama, T.; Miyaji, K.; Sugimoto, M.; Hasegawa, M. Ultrastructure of the dermal chromatophores in a lizard (Scincidae: Plestiodon latiscutatus) with conspicuous body and tail coloration. Zool. Sci. 2006, 23, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Teyssier, J.; Saenko, S.V.; van der Marel, D.; Milinkovitch, M.C. Photonic crystals cause active colour change in chameleons. Nat. Commun. 2015, 6, 6368. [Google Scholar] [CrossRef] [PubMed]
- Posso-Terranova, A.; Andrés, J.Á. Diversification and convergence of aposematic phenotypes: Truncated receptors and cellular arrangements mediate rapid evolution of coloration in harlequin poison frogs. Evol. Int. J. Org. Evol. 2017, 71, 2677–2692. [Google Scholar] [CrossRef]
- Saenko, S.V.; Lamichhaney, S.; Barrio, A.M.; Rafati, N.; Andersson, L.; Milinkovitch, M.C. Amelanism in the corn snake is associated with the insertion of an LTR-retrotransposon in the OCA2 gene. Sci. Rep. 2015, 5, 17118. [Google Scholar] [CrossRef]
- Wang, P.; Xiong, G.; Zeng, D.; Zhang, J.; Ge, L.; Liu, L.; Wang, X.; Hu, Y. Comparative transcriptome and miRNA analysis of skin pigmentation during embryonic development of Chinese soft-shelled turtle (Pelodiscus sinensis). BMC Genom. 2022, 23, 801. [Google Scholar] [CrossRef]
- Lan, W.-J.; Wang, H.-Y.; Lan, W.; Wang, K.-Y. Geniposide enhances melanogenesis by stem cell factor/c-Kit signalling in norepinephrine-exposed normal human epidermal melanocyte. Basic Clin. Pharmacol. Toxicol. 2008, 103, 88–93. [Google Scholar] [CrossRef]
- Garrido, M.C.; Bastian, B.C. KIT as a therapeutic target in melanoma. J. Investig. Dermatol. 2010, 130, 20–27. [Google Scholar] [CrossRef]
- Mizutani, Y.; Hayashi, N.; Kawashima, M.; Imokawa, G. A single UVB exposure increases the expression of functional KIT in human melanocytes by up-regulating MITF expression through the phosphorylation of p38/CREB. Arch. Dermatol. Res. 2010, 302, 283–294. [Google Scholar] [CrossRef]
- Murisier, F.; Beermann, F. Genetics of pigment cells: Lessons from the tyrosinase gene family. Histol. Histopathol. 2006, 21, 567–578. [Google Scholar] [CrossRef]
- Ekblom, R.; Farrell, L.L.; Lank, D.B.; Burke, T. Gene expression divergence and nucleotide differentiation between males of different color morphs and mating strategies in the ruff. Ecol. Evol. 2012, 2, 2485–2505. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Schartl, M. Evolution of endothelin receptors in vertebrates. General. Comp. Endocrinol. 2014, 209, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Fairbank, P.D.; Lee, C.; Ellis, A.; Hildebrand, J.D.; Gross, J.M.; Wallingford, J.B. Shroom2 (APXL) regulates melanosome biogenesis and localization in the retinal pigment epithelium. Development 2006, 133, 4109–4118. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Le, M.-P.; Cannatella, D.; Wallingford, J.B. Changes in localization and expression levels of Shroom2 and spectrin contribute to variation in amphibian egg pigmentation patterns. Dev. Genes Evol. 2009, 219, 319–330. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Schouwey, K.; Beermann, F. The Notch pathway: Hair graying and pigment cell homeostasis. Histol. Histopathol. 2008, 23, 609–619. [Google Scholar] [CrossRef]
- Marks, M.S.; Seabra, M.C. The melanosome: Membrane dynamics in black and white. Nat. Rev. Mol. Cell Biol. 2001, 2, 738–748. [Google Scholar] [CrossRef]
- Raposo, G.; Marks, M.S. Melanosomes—Dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef]
- Fukuda, M.; Kuroda, T.S.; Mikoshiba, K. Slac2-a/melanophilin, the missing link between Rab27 and myosin Va: Implications of a tripartite protein complex for melanosome transport. J. Biol. Chem. 2002, 277, 12432–12436. [Google Scholar] [CrossRef]
- Fukuda, M. Versatile role of Rab27 in membrane trafficking: Focus on the Rab27 effector families. J. Biochem. 2005, 137, 9–16. [Google Scholar] [CrossRef]
- Ohbayashi, N.; Maruta, Y.; Ishida, M.; Fukuda, M. Melanoregulin regulates retrograde melanosome transport through interaction with the RILP–p150Glued complex in melanocytes. J. Cell Sci. 2012, 125, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.A.; Seperack, P.K.; Strobel, M.C.; Copeland, N.G.; Jenkins, N.A. Novel myosin heavy chain encoded by murine dilute coat colour locus. Nature 1991, 349, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Rout, A.K.; Wu, X.; Starich, M.R.; Strub, M.P.; Hammer, J.A.; Tjandra, N. The Structure of Melanoregulin Reveals a Role for Cholesterol Recognition in the Protein’s Ability to Promote Dynein Function. Structure 2018, 26, 1373–1383.e4. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.N.; Wu, X.S.; Rachel, R.A.; Huang, J.D.; Swing, D.A.; Matesic, L.E.; Hammer, J.A., 3rd; Copeland, N.G.; Jenkins, N.A. dsu functions in a MYO5A-independent pathway to suppress the coat color of dilute mice. Proc. Natl. Acad. Sci. USA 2004, 101, 16831–16836. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group 1 | RO Trinity | RU Trinity | RO Protein Coding | RU Protein Coding |
|---|---|---|---|---|
| C | 92.01% | 91.68% | 91.68% | 91.32% |
| S | 24.78% | 23.52% | 24.75% | 23.43% |
| D | 67.23% | 68.16% | 66.94% | 67.89% |
| F | 4.03% | 4.38% | 3.79% | 4.20% |
| I | 0.09% | 0.00% | 0.09% | 0.03% |
| M | 3.88% | 3.94% | 4.44% | 4.44% |
| Database | RO | RU |
|---|---|---|
| Iprscan | 28,860 | 25,522 |
| GO | 18,561 | 16,516 |
| KEGG | 13,499 | 12,723 |
| NR | 22,319 | 20,961 |
| SwissProt | 15,737 | 15,018 |
| Annotated | 30,637 | 27,281 |
| Unannotated | 33,572 | 33,571 |
| Total | 64,209 | 60,852 |
| GeneID | Group | LogFC | p-Value | FDR |
|---|---|---|---|---|
| Unigene9453 (shroom2) | ROB_vs_ROP | −5.410569297 | 1.02 × 10−6 | 0 |
| Unigene11641 (shroom2) | ROB_vs_ROP | −3.182307072 | 5.68 × 10−6 | 0 |
| Unigene9433 (shroom2) | ROB_vs_ROP | −4.081419141 | 3.39 × 10−5 | 0 |
| Unigene9417 (notch1) | ROB_vs_ROP | −4.479233533 | 8.00 × 10−5 | 0.01 |
| GroupID | GeneName | Counts | Mutation |
|---|---|---|---|
| Cluster1019 | MC1R | 1 | V144A |
| Cluster1020 | MC1R | 1 | S135N |
| Cluster1147 | shroom2 | 2 | T389P, N1086T |
| Cluster1151 | shroom2 | 5 | E586D, N637S, E660K, T756A, T776M |
| Cluster1193 | adam17 | 1 | D26E |
| Cluster1197 | notch1 | 1 | H90R |
| Cluster1198 | shroom2 | 1 | S530P |
| Cluster1287 | MC1R | 1 | V133I |
| Cluster1312 | notch1 | 1 | H27Q |
| Cluster1333 | MC1R | 1 | A101V |
| Cluster1404 | mreg | 1 | V14A |
| Cluster1428 | MC1R | 1 | L42F |
| Cluster1437 | MC1R | 1 | S69P |
| Cluster1455 | notch1 | 1 | N749S |
| Cluster797 | KIT | 1 | P85A |
| Cluster877 | notch1 | 1 | E21A |
| Cluster896 | ednrb | 2 | Y42F, D68N |
| Cluster897 | ednrb | 1 | K179R |
| Cluster928 | ednrb | 2 | L202F, L307V |
| Cluster941 | notch1 | 1 | Y25H |
| Cluster958 | MC1R | 1 | H46Q |
| Cluster980 | MC1R | 1 | L226S |
| GeneID | Sample A | Sample B | logFC | Log Counts per Million (logCPM) | Probability Value (p-Value) | FDR |
|---|---|---|---|---|---|---|
| RU_DN1145_c3_g2 (mreg) | ROP | RUR | −15.19373837 | 5.77320102074195 | 1.3015005339553 × 10−36 | 1.38737854091305 × 10−35 |
| RU_DN10511_c0_g1 (notch1) | ROP | RUR | −10.82609391 | 1.42891518860247 | 1.30325616007406 × 10−27 | 7.40272367248823 × 10−27 |
| Unigene11172 (notch1) | ROP | RUR | 2.26713514896129 | 1.73494640766586 | 5.02842825852524 × 10−5 | 7.21132394782175 × 10−5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, K.; Lu, A.; Xu, Y.; Zhu, F. Comparison of Transcriptome Differences in Scales of Two Closely Related Snake Species (Lycodon rufozonatus and Lycodon rosozonatus). Animals 2025, 15, 1061. https://doi.org/10.3390/ani15071061
Sun K, Lu A, Xu Y, Zhu F. Comparison of Transcriptome Differences in Scales of Two Closely Related Snake Species (Lycodon rufozonatus and Lycodon rosozonatus). Animals. 2025; 15(7):1061. https://doi.org/10.3390/ani15071061
Chicago/Turabian StyleSun, Ke, Anqiong Lu, Yu Xu, and Fei Zhu. 2025. "Comparison of Transcriptome Differences in Scales of Two Closely Related Snake Species (Lycodon rufozonatus and Lycodon rosozonatus)" Animals 15, no. 7: 1061. https://doi.org/10.3390/ani15071061
APA StyleSun, K., Lu, A., Xu, Y., & Zhu, F. (2025). Comparison of Transcriptome Differences in Scales of Two Closely Related Snake Species (Lycodon rufozonatus and Lycodon rosozonatus). Animals, 15(7), 1061. https://doi.org/10.3390/ani15071061