
Chromosome-Level Genome Assembly of the Meishan Pig and Insights into Its Domestication Mechanisms

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Sequencing of the Genome
2.3. Genome Size Estimation and De Novo Assembly
2.4. Genome Quality Assessment
2.5. Distribution of Repetitive Elements and Genome Annotation
2.6. Genomic Structural Variation Analysis
2.7. Domestication Signal Analysis of Meishan Pigs
3. Results
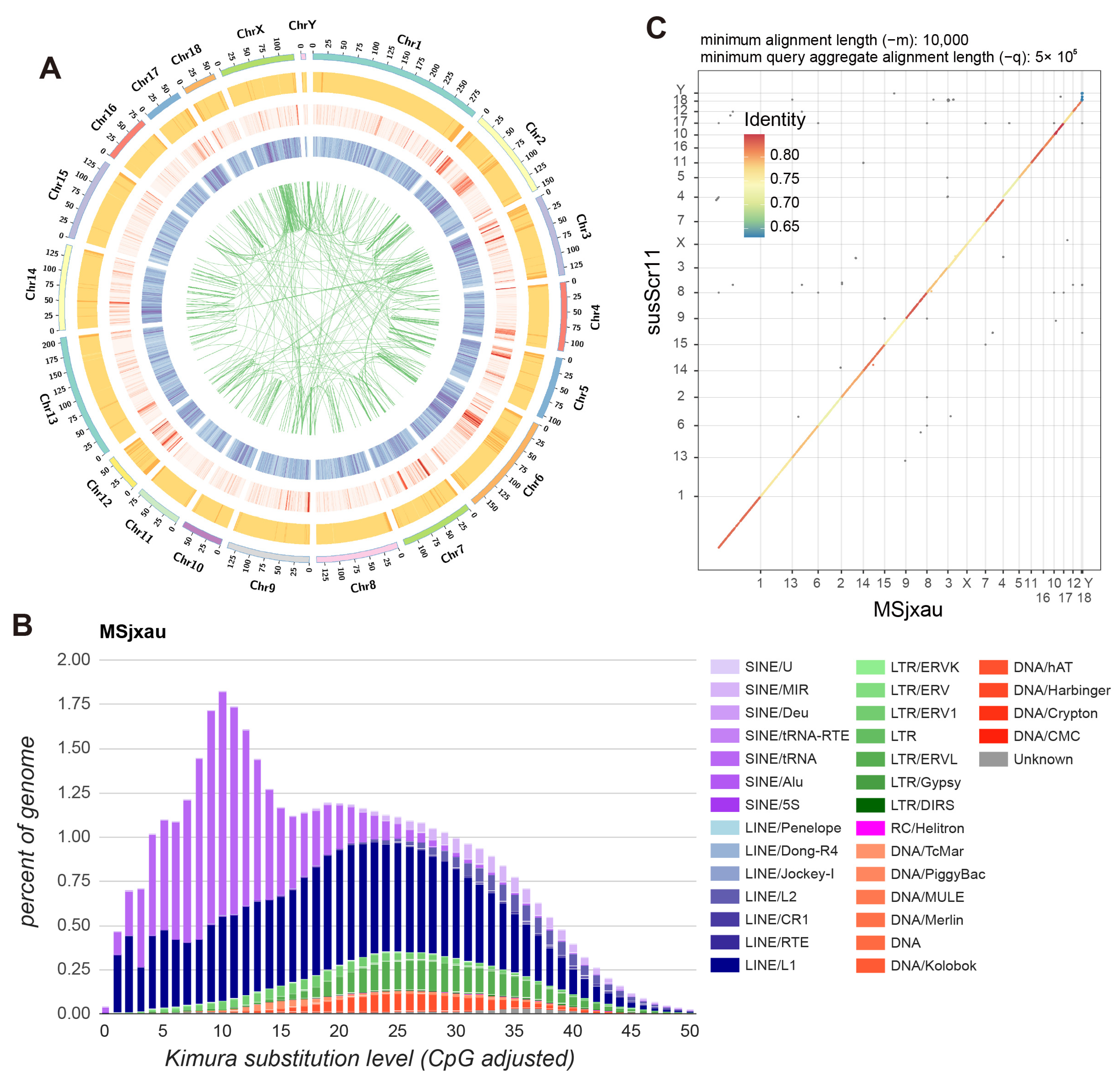
3.1. Genome Assembly and Evaluation of the Meishan Pig
3.2. Genome Annotation of the Meishan Pig
3.3. Comparison of Genomic Structural Variation
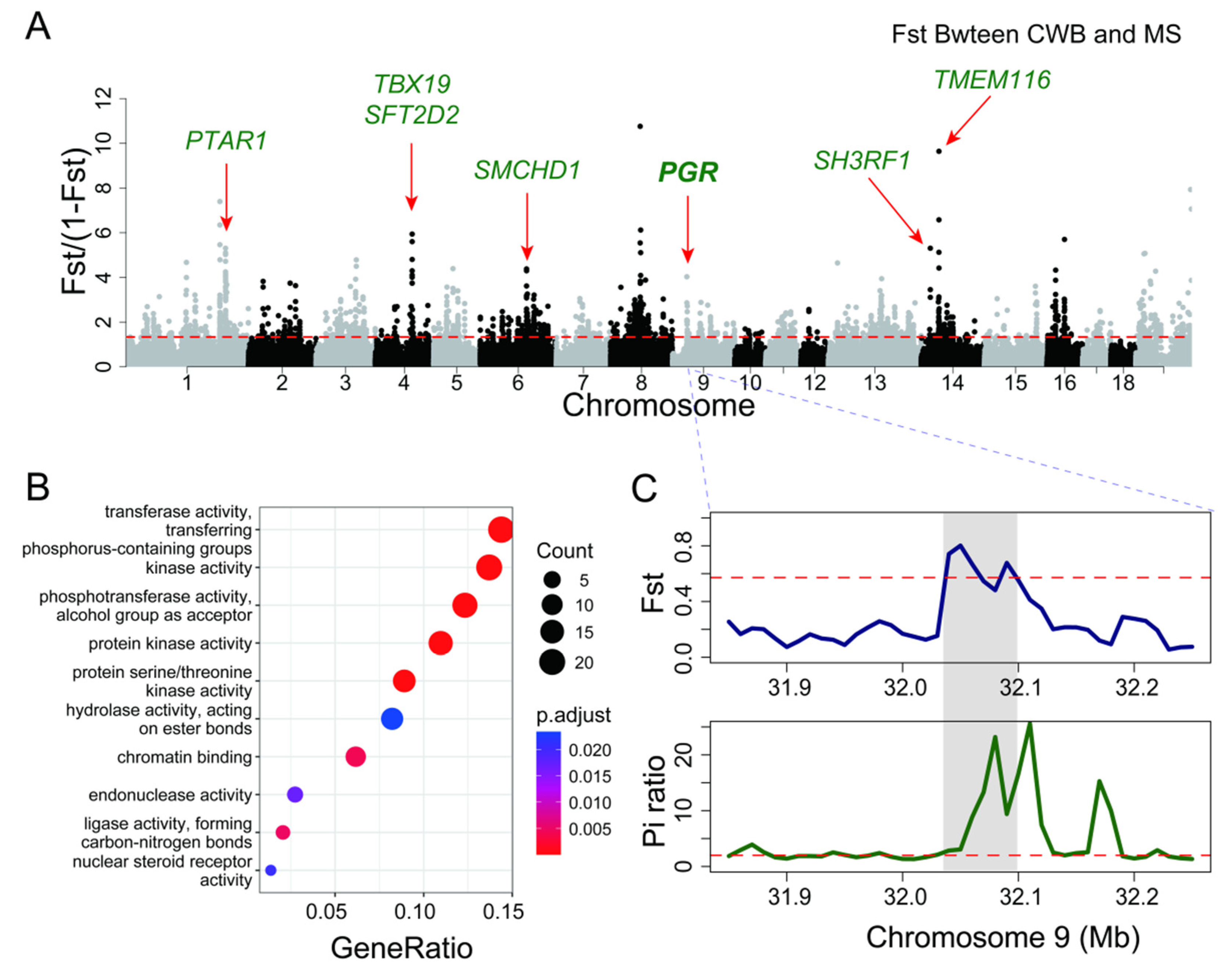
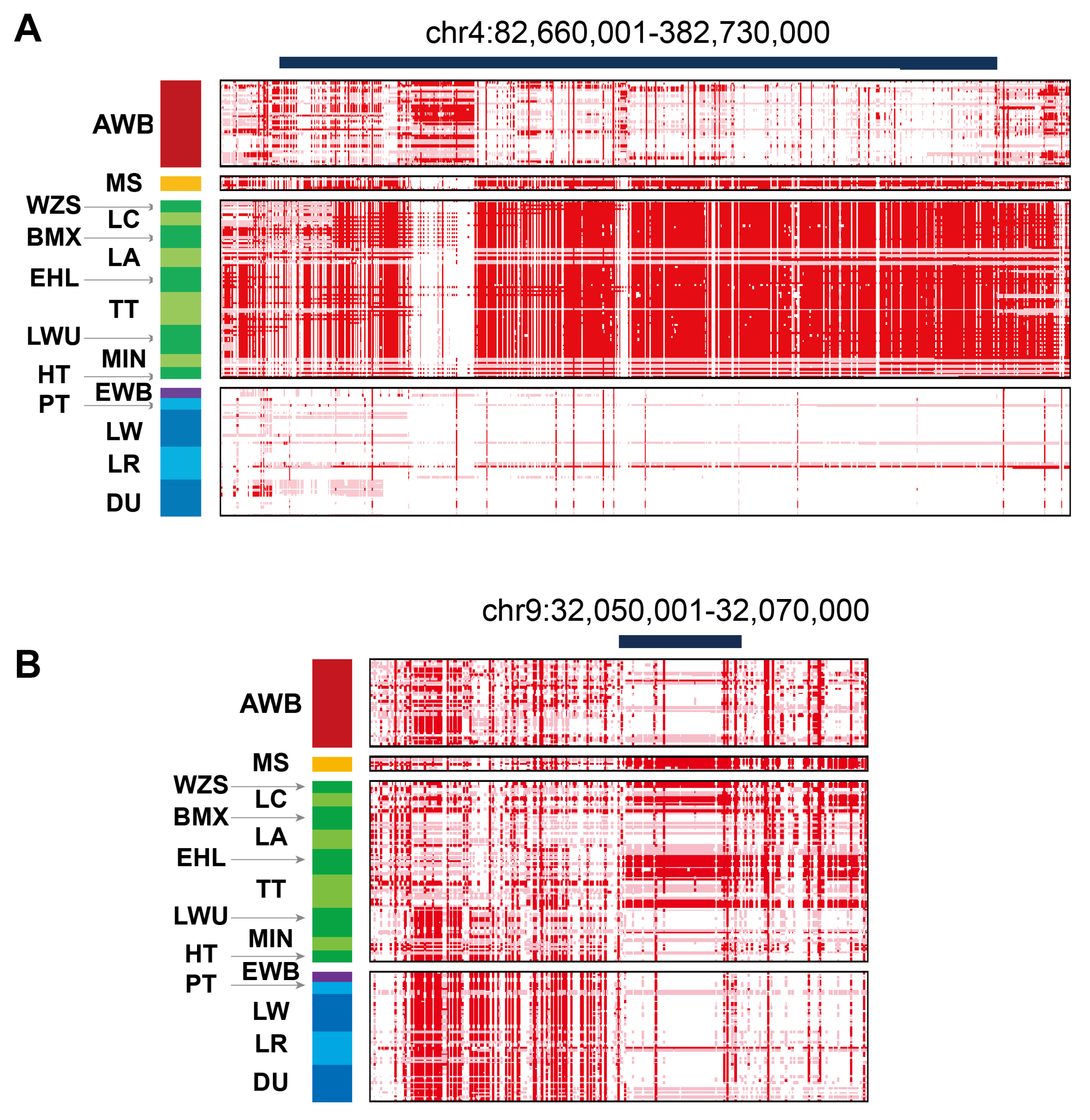
3.4. The Selective Domestication Between Chinese Wild Boar and Meishan Pig
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warr, A.; Affara, N.; Aken, B.; Beiki, H.; Bickhart, D.M.; Billis, K.; Chow, W.; Eory, L.; Finlayson, H.A.; Flicek, P. An improved pig reference genome sequence to enable pig genetics and genomics research. Gigascience 2020, 9, giaa051. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.-S.; Li, M.; Chen, T.; Wei, X.-C.; Qi, Q.-E.; Cheng, X.; Li, C.-Y.; Jiang, Q.-Y.; Xi, Q.-Y.; Zhang, Y.-L. miRNAome, mRNAome and degradome analysis of Tibetan minipigs anterior pituitary. Gen. Comp. Endocrinol. 2018, 259, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Zak, L.J.; Gaustad, A.H.; Bolarin, A.; Broekhuijse, M.L.; Walling, G.A.; Knol, E.F. Genetic control of complex traits, with a focus on reproduction in pigs. Mol. Reprod. Dev. 2017, 84, 1004–1011. [Google Scholar] [CrossRef]
- Chen, K.; Baxter, T.; Muir, W.M.; Groenen, M.A.; Schook, L.B. Genetic resources, genome mapping and evolutionary genomics of the pig (Sus scrofa). Int. J. Biol. Sci. 2007, 3, 153. [Google Scholar] [CrossRef]
- Tong, X.; Chen, D.; Hu, J.; Lin, S.; Ling, Z.; Ai, H.; Zhang, Z.; Huang, L. Accurate haplotype construction and detection of selection signatures enabled by high quality pig genome sequences. Nat. Commun. 2023, 14, 5126. [Google Scholar] [CrossRef]
- Du, H.; Zhou, L.; Liu, Z.; Zhuo, Y.; Zhang, M.; Huang, Q.; Lu, S.; Xing, K.; Jiang, L.; Liu, J.-F. The 1000 Chinese Indigenous Pig Genomes Project provides insights into the genomic architecture of pigs. Nat. Commun. 2024, 15, 10137. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yin, H.; Bai, L.; Yao, W.; Tao, T.; Zhao, Q.; Gao, Y.; Teng, J.; Xu, Z.; Lin, Q. Mapping and functional characterization of structural variation in 1060 pig genomes. Genome Biol. 2024, 25, 116. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wu, J.; Huang, X.; Zhou, Y.; Zhang, Y.; Liu, M.; Liu, Q.; Ke, S.; He, M.; Fu, H. ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs. Nature 2022, 606, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, J.; Zhou, L.; Ren, J.; Liu, X.; Zhang, H.; Yang, B.; Zhang, Z.; Ma, H.; Xie, X. A splice mutation in the PHKG1 gene causes high glycogen content and low meat quality in pig skeletal muscle. PLoS Genet. 2014, 10, e1004710. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.D.; Wagner, J.; Dwarshuis, N.; Miga, K.H.; Sedlazeck, F.J.; Salit, M.; Zook, J.M. Variant calling and benchmarking in an era of complete human genome sequences. Nat. Rev. Genet. 2023, 24, 464–483. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Gong, H.; Zhou, Z.; Jiang, T.; Lin, Z.; Li, J.; Xiao, S.; Yang, B.; Huang, L. Mapping short tandem repeats for liver gene expression traits helps prioritize potential causal variants for complex traits in pigs. J. Anim. Sci. Biotechnol. 2022, 13, 8. [Google Scholar] [CrossRef]
- Espinosa, E.; Bautista, R.; Larrosa, R.; Plata, O. Advancements in long-read genome sequencing technologies and algorithms. Genomics 2024, 116, 110842. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Du, H.; Lu, S.; Huang, Q.; Zhou, L.; Liu, J.-F. Chromosome-level genome assembly of Huai pig (Sus scrofa). Sci. Data 2024, 11, 1072. [Google Scholar] [CrossRef] [PubMed]
- Wy, S.; Kwon, D.; Park, W.; Chai, H.-H.; Cho, I.-C.; Kim, J. A chromosome-level genome assembly of the Korean minipig (Sus scrofa). Sci. Data 2024, 11, 840. [Google Scholar] [CrossRef]
- Cao, C.; Miao, J.; Xie, Q.; Sun, J.; Cheng, H.; Zhang, Z.; Wu, F.; Liu, S.; Ye, X.; Zhang, Z. The first near-complete genome assembly of pig: Enabling more accurate genetic research. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, Y.; Wang, M.; Guo, Y.; Liang, J.; Yang, X.; Qi, W.; Wu, Y.; Si, J.; Zhu, S. Development and genome sequencing of a laboratory-inbred miniature pig facilitates study of human diabetic disease. iScience 2019, 19, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rhie, A.; Hansen, N.F.; Koren, S.; Phillippy, A.M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 2022, 19, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fu, Y.; Yang, Y.; Yi, G.; Lian, J.; Xie, B.; Yao, Y.; Chen, M.; Niu, Y.; Liu, L. Integration of multi-omics data reveals cis-regulatory variants that are associated with phenotypic differentiation of eastern from western pigs. Genet. Sel. Evol. 2022, 54, 62. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Park, N.; Wy, S.; Lee, D.; Chai, H.-H.; Cho, I.-C.; Lee, J.; Kwon, K.; Kim, H.; Moon, Y. A chromosome-level genome assembly of the Korean crossbred pig Nanchukmacdon (Sus scrofa). Sci. Data 2023, 10, 761. [Google Scholar] [CrossRef] [PubMed]
- Gabur, I.; Chawla, H.S.; Snowdon, R.J.; Parkin, I.A. Connecting genome structural variation with complex traits in crop plants. Theor. Appl. Genet. 2019, 132, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fang, J.; Wang, Y.; Wang, P.; Zhang, S.; Liao, Z.; Lu, H.; Zhang, X. Evolutionary genomics of structural variation in the tea plant, Camellia sinensis. Trop. Plants 2022, 1, 1–11. [Google Scholar] [CrossRef]
- Liao, W.-W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J. A draft human pangenome reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]
- Yang, B.; Cui, L.; Perez-Enciso, M.; Traspov, A.; Crooijmans, R.P.; Zinovieva, N.; Schook, L.B.; Archibald, A.; Gatphayak, K.; Knorr, C. Genome-wide SNP data unveils the globalization of domesticated pigs. Genet. Sel. Evol. 2017, 49, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Li, S.T.; Yao, W.Y.; Xie, C.D.; Chen, Z.; Zeng, Z.J.; Wang, D.; Xu, K.; Shen, Z.J.; Mu, Y. The Meishan pig genome reveals structural variation-mediated gene expression and phenotypic divergence underlying Asian pig domestication. Mol. Ecol. Resour. 2021, 21, 2077–2092. [Google Scholar] [CrossRef] [PubMed]
- Albarella, U.; Dobney, K.; Rowley-Conwy, P. The Domestication of the Pig (Sus scrofa): New Challenges and Approaches; University of California Press: Oakland, CA, USA, 2006. [Google Scholar]
- Wehr, N.H. Historical range expansion and biological changes of Sus scrofa corresponding to domestication and feralization. Mammal Res. 2021, 66, 1–12. [Google Scholar] [CrossRef]
- Zhao, P.; Yu, Y.; Feng, W.; Du, H.; Yu, J.; Kang, H.; Zheng, X.; Wang, Z.; Liu, G.E.; Ernst, C.W. Evidence of evolutionary history and selective sweeps in the genome of Meishan pig reveals its genetic and phenotypic characterization. Gigascience 2018, 7, giy058. [Google Scholar] [CrossRef]
- Collingbourne, S.J. Conservation Genetics of Traditional and Commercial Pig Breeds, and Evaluation of Their Crossbreeding Potential for Productivity Improvement; University of Essex: Wivenhoe Park, UK, 2019. [Google Scholar]
- Gong, Y.; Li, Y.; Liu, X.; Ma, Y.; Jiang, L. A review of the pangenome: How it affects our understanding of genomic variation, selection and breeding in domestic animals? J. Anim. Sci. Biotechnol. 2023, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.S.; Urban, A.E.; Mills, R.E. Structural variation in the sequencing era. Nat. Rev. Genet. 2020, 21, 171–189. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Hastie, A.R.; Cao, D.; Lam, E.T.; Sun, Y.; Huang, H.; Liu, X.; Lin, L.; Andrews, W.; Chan, S. Rapid detection of structural variation in a human genome using nanochannel-based genome mapping technology. GigaScience 2014, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; Weissensteiner, M.H.; Sedlazeck, F.J. Towards population-scale long-read sequencing. Nat. Rev. Genet. 2021, 22, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Eggertsson, H.P.; Kristmundsdottir, S.; Beyter, D.; Jonsson, H.; Skuladottir, A.; Hardarson, M.T.; Gudbjartsson, D.F.; Stefansson, K.; Halldorsson, B.V.; Melsted, P. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat. Commun. 2019, 10, 5402. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Krusche, P.; Dolzhenko, E.; Sherman, R.M.; Petrovski, R.; Schlesinger, F.; Kirsche, M.; Bentley, D.R.; Schatz, M.C.; Sedlazeck, F.J. Paragraph: A graph-based structural variant genotyper for short-read sequence data. Genome Biol. 2019, 20, 291. [Google Scholar] [CrossRef]
- Du, H.; Zhuo, Y.; Lu, S.; Li, W.; Zhou, L.; Sun, F.; Liu, G.; Liu, J.-F. Pangenome Reveals Gene Content Variations and Structural Variants Contributing to Pig Characteristics. Genom. Proteom. Bioinform. 2024, qzae081. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Długosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Freire, B.; Ladra, S.; Paramá, J.R. Memory-efficient assembly using Flye. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 19, 3564–3577. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Xu, M.; Guo, L.; Gu, S.; Wang, O.; Zhang, R.; Peters, B.A.; Fan, G.; Liu, X.; Xu, X.; Deng, L. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 2020, 9, giaa094. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Han, D. BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics 2022, 38, 2404–2413. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2019; pp. 227–245. [Google Scholar]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef]
- Luu, P.-L.; Ong, P.-T.; Dinh, T.-P.; Clark, S.J. Benchmark study comparing liftover tools for genome conversion of epigenome sequencing data. NAR Genom. Bioinform. 2020, 2, lqaa054. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.-F.; Wang, S.; Wang, C.-L.; Xu, R.-H.; Wang, W.-W.; Jiang, Y.; Wang, M.-S.; Jiang, L.; Dai, L.-H.; Wang, J.-R. Pangenome obtained by long-read sequencing of 11 genomes reveal hidden functional structural variants in pigs. iScience 2023, 26, 106119. [Google Scholar] [CrossRef]
- Niu, Y.; Fan, X.; Yang, Y.; Li, J.; Lian, J.; Wang, L.; Zhang, Y.; Tang, Y.; Tang, Z. Haplotype-resolved assembly of a pig genome using single-sperm sequencing. Commun. Biol. 2024, 7, 738. [Google Scholar] [CrossRef]
- Ma, H.; Jiang, J.; He, J.; Liu, H.; Han, L.; Gong, Y.; Li, B.; Yu, Z.; Tang, S.; Zhang, Y. Long-read assembly of the Chinese indigenous Ningxiang pig genome and identification of genetic variations in fat metabolism among different breeds. Mol. Ecol. Resour. 2022, 22, 1508–1520. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, G.; Liu, X.; Skovgaard, K.; Sturek, M.; Heegaard, P.M. The genome of the naturally evolved obesity-prone Ossabaw miniature pig. iScience 2021, 24, 103081. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.-B.; Yan, C.; Adeola, A.C.; Wang, K.; Huang, C.-P.; Xu, M.-M.; Qiu, Q.; Yin, X.; Fan, C.-Y.; Ma, Y.-F. African suid genomes provide insights into the local adaptation to diverse African environments. Mol. Biol. Evol. 2022, 39, msac256. [Google Scholar] [CrossRef]
- Wang, Y.; Gou, Y.; Yuan, R.; Zou, Q.; Zhang, X.; Zheng, T.; Fei, K.; Shi, R.; Zhang, M.; Li, Y. A chromosome-level genome of Chenghua pig provides new insights into the domestication and local adaptation of pigs. Int. J. Biol. Macromol. 2024, 270, 131796. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, X.; Chu, C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 2020, 21, 265. [Google Scholar] [CrossRef] [PubMed]
- Kirsche, M.; Prabhu, G.; Sherman, R.; Ni, B.; Aganezov, S.; Schatz, M.C. Jasmine: Population-scale structural variant comparison and analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Li, X.; Liu, Q.; Fu, C.; Li, M.; Li, C.; Li, X.; Zhao, S.; Zheng, Z. Characterizing structural variants based on graph-genotyping provides insights into pig domestication and local adaption. J. Genet. Genom. 2024, 51, 394–406. [Google Scholar] [CrossRef]
- Khan, M.M.H.; Rafii, M.Y.; Ramlee, S.I.; Jusoh, M.; Al Mamun, M.; Halidu, J. DNA fingerprinting, fixation-index (Fst), and admixture mapping of selected Bambara groundnut (Vigna subterranea [L.] Verdc.) accessions using ISSR markers system. Sci. Rep. 2021, 11, 14527. [Google Scholar]
- Zhu, Y.; Li, W.; Yang, B.; Zhang, Z.; Ai, H.; Ren, J.; Huang, L. Signatures of selection and interspecies introgression in the genome of Chinese domestic pigs. Genome Biol. Evol. 2017, 9, 2592–2603. [Google Scholar] [CrossRef]
- Dinh, D.T.; Breen, J.; Nicol, B.; Foot, N.J.; Bersten, D.C.; Emery, A.; Smith, K.M.; Wong, Y.Y.; Barry, S.C.; Yao, H.H. Progesterone receptor mediates ovulatory transcription through RUNX transcription factor interactions and chromatin remodelling. Nucleic Acids Res. 2023, 51, 5981–5996. [Google Scholar] [CrossRef]
- Zeberg, H.; Kelso, J.; Pääbo, S. The Neandertal progesterone receptor. Mol. Biol. Evol. 2020, 37, 2655–2660. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fu, J.; Wang, A. Expression of genes involved in progesterone receptor paracrine signaling and their effect on litter size in pigs. J. Anim. Sci. Biotechnol. 2016, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Xiao, X.; Zhang, Z.; Li, M. Genetic insights and neurobiological implications from NRXN1 in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1400–1414. [Google Scholar] [CrossRef]
- Bonnet, A.; Le Cao, K.-A.; Sancristobal, M.; Benne, F.; Robert-Granié, C.; Law-So, G.; Fabre, S.; Besse, P.; De Billy, E.; Quesnel, H. In vivo gene expression in granulosa cells during pig terminal follicular development. Reproduction 2008, 136, 211–224. [Google Scholar] [CrossRef]
- Massah, S.; Jubene, J.; Lee, F.; Beischlag, T.; Prefontaine, G. The effects of the DNA Demethylating reagent, 5-azacytidine on SMCHD1 genomic localization. BMC Genet. 2020, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Kang, Y.K.; Park, Z.Y.; Kim, Y.-H.; Hong, S.W.; Oh, S.J.; Sohn, H.A.; Yang, S.-J.; Jang, Y.J.; Lee, D.C. SH3RF2 functions as an oncogene by mediating PAK4 protein stability. Carcinogenesis 2014, 35, 624–634. [Google Scholar] [CrossRef]
- Wesoly, J.; Pstrąg, N.; Derylo, K.; Michalec-Wawiórka, B.; Derebecka, N.; Nowicka, H.; Kajdasz, A.; Kluzek, K.; Srebniak, M.; Tchórzewski, M. Structural, topological, and functional characterization of transmembrane proteins TMEM213, 207, 116, 72 and 30B provides a potential link to ccRCC etiology. Am. J. Cancer Res. 2023, 13, 1863. [Google Scholar]
- Tong, S.-f.; Mo, Z.; Rui, X.; Li, D.-f.; Zhang, L.-f.; Yang, L. Genome-wide detection for runs of homozygosity analysis in three pig breeds from Chinese Taihu Basin and Landrace pigs by SLAF-seq data. J. Integr. Agric. 2022, 21, 3293–3301. [Google Scholar] [CrossRef]
- Lander, B.; Schneider, M.; Brunson, K. A history of pigs in China: From curious omnivores to industrial pork. J. Asian Stud. 2020, 79, 865–889. [Google Scholar] [CrossRef]
- Wu, H.; Luo, L.-Y.; Zhang, Y.-H.; Zhang, C.-Y.; Huang, J.-H.; Mo, D.-X.; Zhao, L.-M.; Wang, Z.-X.; Wang, Y.-C.; He-Hua, E. Telomere-to-telomere genome assembly of a male goat reveals variants associated with cashmere traits. Nat. Commun. 2024, 15, 10041. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Q.; Hu, Y.; Yu, Y.; Zheng, K.; Li, D.; Qin, L.; Yu, X. The complete telomere-to-telomere sequence of a mouse genome. Science 2024, 386, 1141–1146. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly | MSjxau | Sscrofa11.1 |
|---|---|---|
| Total length of assembly (Gb) | 2.45 | 2.5 |
| Min length (bp) | 1008 | 15,096 |
| Max length (Mb) | 292.79 | 274.33 |
| Number of scaffolds | 1144 | 613 |
| Number of gaps | 466 | 544 |
| Quality values (QV) | 37.06 | NA |
| Scaffold N50 (Mb) | 139.17 | 138.97 |
| Contig N50 (Mb) | 15.62 | 48.23 |
| Assembly | Groups | Levels | Assembly | Groups | Levels |
|---|---|---|---|---|---|
| PHA_SAMN15809474 | Outgroup | Chromosome | LC_CNA0017742 | Asian | Contig |
| PHA_SAMC146517 | Outgroup | Chromosome | TT_CRR307872 | Asian | Contig |
| POT_SAMC146518 | Outgroup | Chromosome | MS_CRR307869 | Asian | Contig |
| BMX_SAMN09531794 | Asian | Chromosome | RC_CRR307871 | Asian | Contig |
| DNXE_SAMC146519 | Asian | Chromosome | LWU_CRR307868 | Asian | Contig |
| MS_SAMN14422843 | Asian | Chromosome | AQLB_CRR307865 | Asian | Contig |
| MS_jxau | Asian | Chromosome | MIN_CRR307870 | Asian | Contig |
| SWT_PRJCA017882 | Asian | Chromosome | OSS_SAMN28857594 | European | Chromosome |
| TC_PRJCA017882 | Asian | Chromosome | DU_SAMN15501053 | European | Chromosome |
| NX_SAMN12123497 | Asian | Chromosome | DLY_SAMN07325927 | European | Chromosome |
| HUAI_SAMN43144123 | Asian | Chromosome | BBH_SAMN37151398 | European | Chromosome |
| CH_SAMN36411884 | Asian | Chromosome | LR_SRR24831861 | European | Contig |
| KORE_SAMN41069951 | Asian | Chromosome | LW_CRR307874 | European | Contig |
| NANC_SAMN34578002 | Asian | Chromosome | LW_CRR307874 | European | Contig |
| WZS_CRR307873 | Asian | Contig |
| Region | Gene | Top Fst | Description |
|---|---|---|---|
| chr1:223030001-223100000 | PTAR1 | 0.841 | Protein prenyltransferase activity |
| chr3:90190001-90220000 | NRXN1 | 0.827 | Nervous development |
| chr4:82660001-82730000 | TBX19 | 0.848 | Developmental processes |
| chr4:82660001-82730000 | SFT2D2 | 0.856 | lipid metabolism |
| chr6:103930001-103970000 | SMCHD1 | 0.814 | Epigenetic silencing |
| chr9:32050001-32070000 | PGR | 0.801 | Progesterone Receptor |
| chr14:20450001-20470000 | SH3RF1 | 0.841 | Protein ligase activity |
| chr14:39710001-39780000 | TMEM116 | 0.906 | Transmembrane protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, H.; Hu, J.; Zhang, Z.; Wu, Z. Chromosome-Level Genome Assembly of the Meishan Pig and Insights into Its Domestication Mechanisms. Animals 2025, 15, 603. https://doi.org/10.3390/ani15040603
Du H, Hu J, Zhang Z, Wu Z. Chromosome-Level Genome Assembly of the Meishan Pig and Insights into Its Domestication Mechanisms. Animals. 2025; 15(4):603. https://doi.org/10.3390/ani15040603
Chicago/Turabian StyleDu, Huipeng, Jianchao Hu, Zhiyan Zhang, and Zhongzi Wu. 2025. "Chromosome-Level Genome Assembly of the Meishan Pig and Insights into Its Domestication Mechanisms" Animals 15, no. 4: 603. https://doi.org/10.3390/ani15040603
APA StyleDu, H., Hu, J., Zhang, Z., & Wu, Z. (2025). Chromosome-Level Genome Assembly of the Meishan Pig and Insights into Its Domestication Mechanisms. Animals, 15(4), 603. https://doi.org/10.3390/ani15040603