
Genome Sequencing Reveals the Adaptation of Chickens to High Altitudes in Different Regions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Resources
2.2. Genetic Variation Detection
2.3. Phylogenetic Analysis and Principal Component Analysis
2.4. Analysis of Population Genetic Structure
2.5. Dimensionality Reduction Methods Reveal the Fine Structure of Chicken Populations
2.6. Linkage Disequilibrium Decay
2.7. Selective Clearance Analysis
2.8. Runs of Homozygosity Analysis
2.9. Gene Flow Analysis
2.10. Gene Annotation and Enrichment Analysis
3. Results
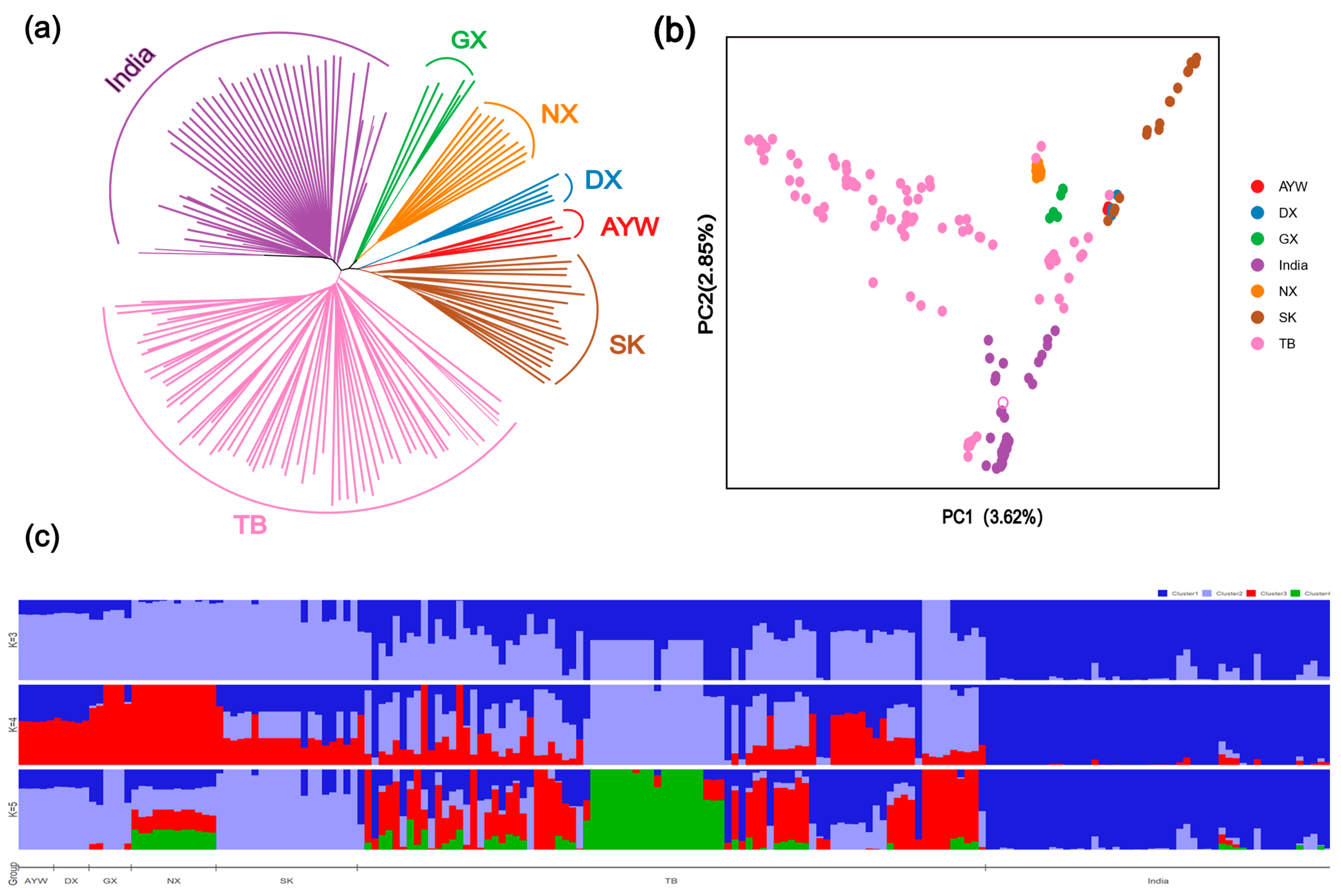
3.1. Analysis of Population Genetic Structure
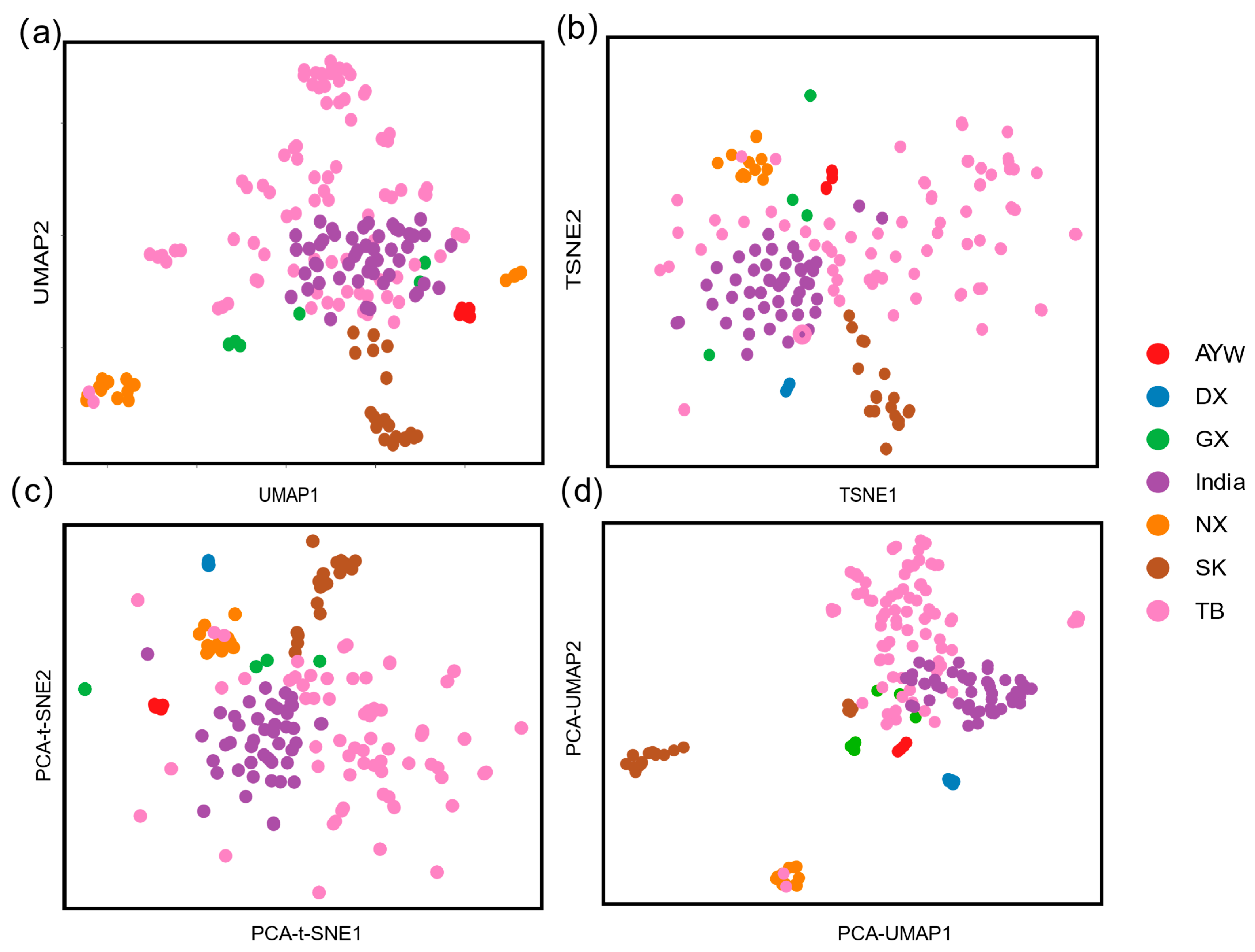
3.2. Different Dimensionality Reduction Methods Reveal the Fine Structure of Chicken Populations
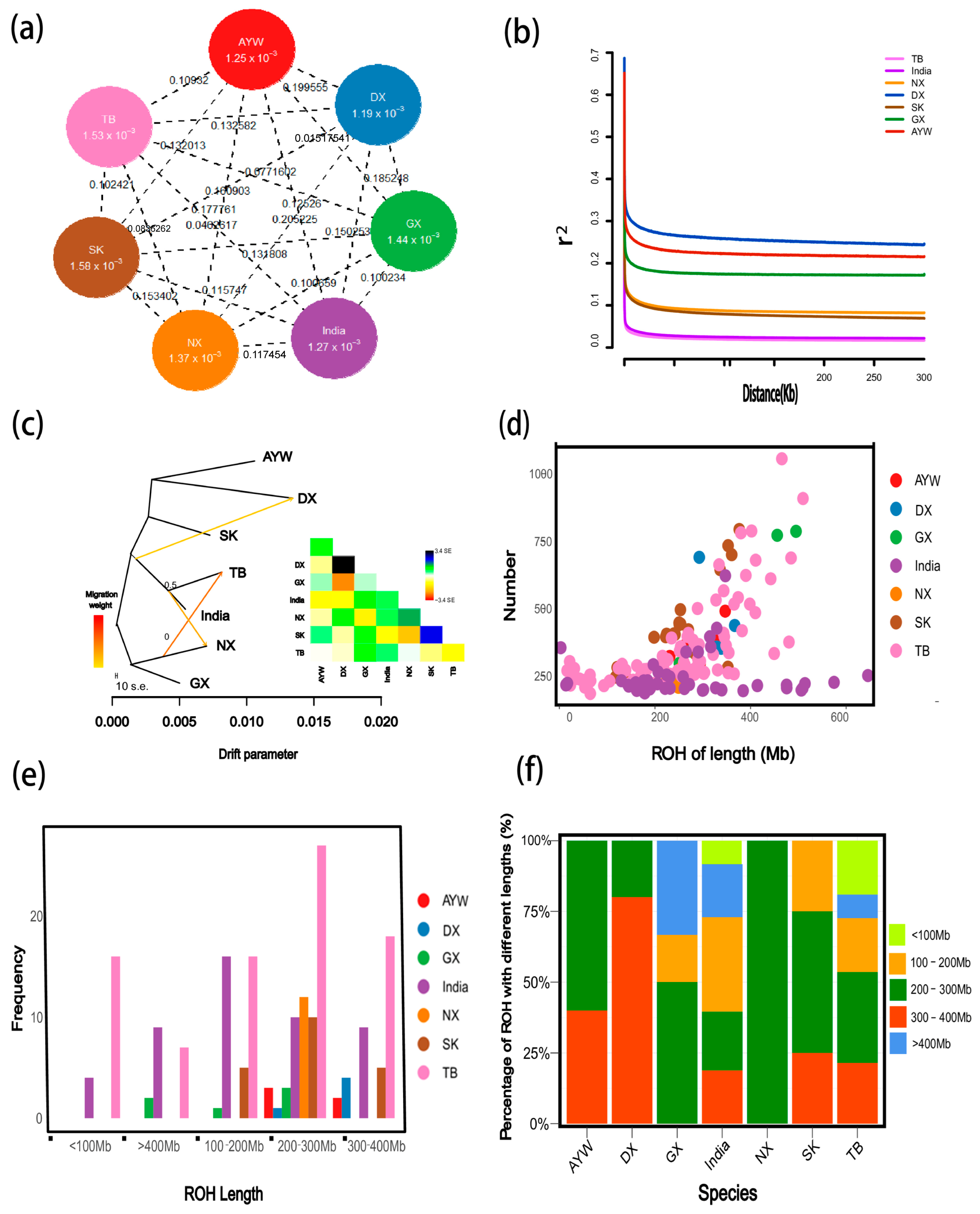
3.3. Genetic Distance and Gene Flow Analysis
3.4. Linkage Disequilibrium Decay
3.5. Runs of Homozygosity
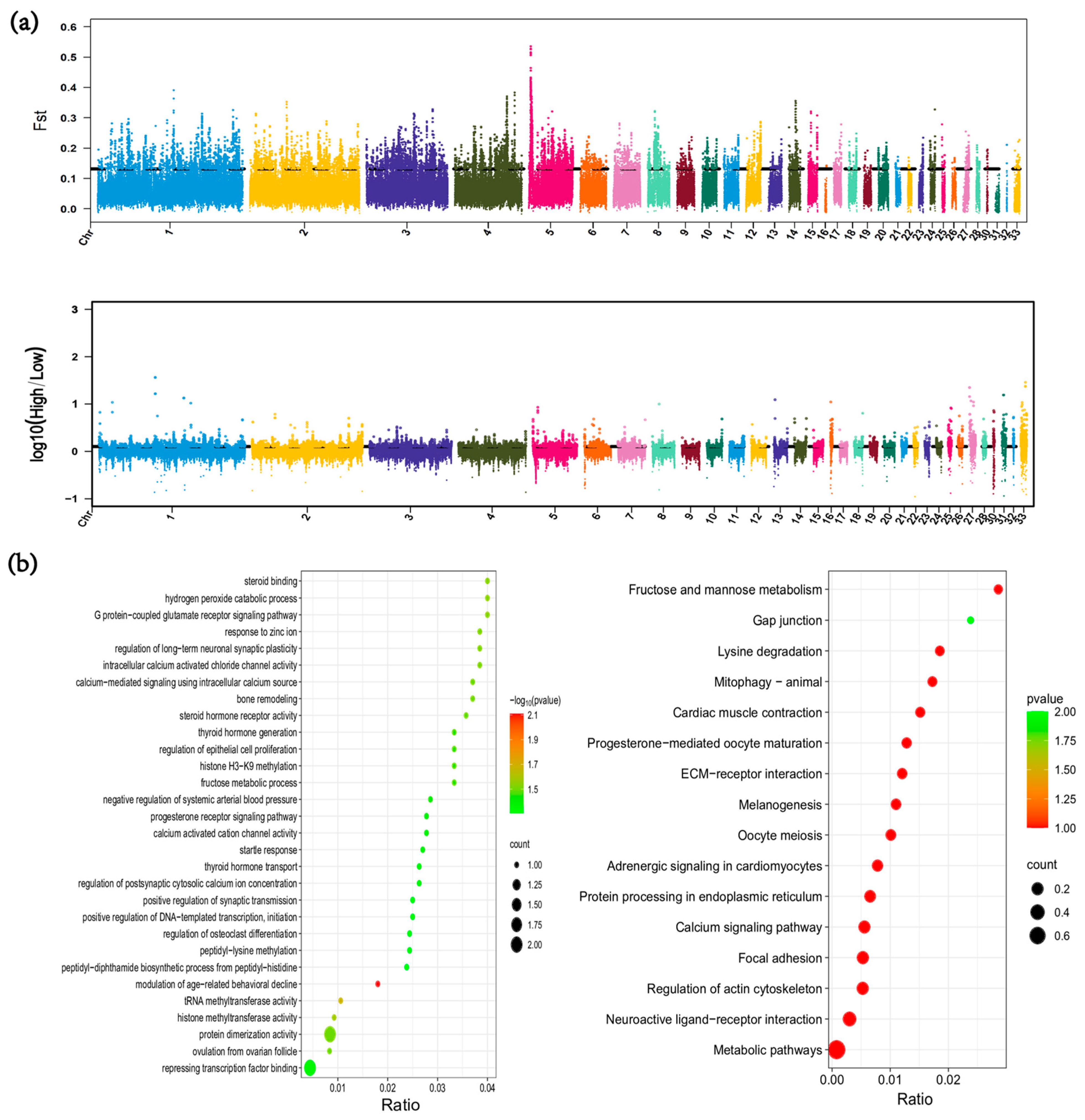
3.6. Genome-Wide Selection Signal Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lawler, A. Why Did the Chicken Cross the World? Atria Books: New York, NY, USA, 2014. [Google Scholar]
- Lawal, R.A.; Hanotte, O. Domestic chicken diversity: Origin, distribution, and adaptation. Anim. Genet. 2021, 52, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-S.; Thakur, M.; Peng, M.-S.; Jiang, Y.; Frantz, L.A.F.; Li, M.; Zhang, J.-J.; Wang, S.; Peters, J.; Otecko, N.O.; et al. 863 genomes reveal the origin and domestication of chicken. Cell Res. 2020, 30, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Bi, D.; Xiao, H.; Zhou, C.; Zhou, J.; Deng, X.; Liu, X. Systems Biology Approach to Study the High Altitude Adaptation in Tibetans. Braz. Arch. Biol. Technol. 2013, 56, 53–60. [Google Scholar] [CrossRef]
- Guang, X.E.; Yang, B.G.; Zhu, Y.B.; Duang, X.H.; Basang, W.D.; Luo, X.L.; An, T.W. Genome-wide selective sweep analysis of the high-altitude adaptability of yaks by using the copy number variant. 3 Biotech 2020, 10, 259. [Google Scholar] [CrossRef]
- Edea, Z.; Dadi, H.; Dessie, T.; Kim, K.-S. Genomic signatures of high-altitude adaptation in Ethiopian sheep populations. Genes Genom. 2019, 41, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Liu, Y.; Zhang, R.; Zheng, X.; Zhao, G.; Li, F.; Liu, W.; Yue, B.; Yang, N. Chromosome-level Genome Assembly of the High-altitude Leopard (Panthera pardus) Sheds Light on Its Environmental Adaptation. Genome Biol. Evol. 2022, 14, evac128. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, S.; Yang, Y.; Tan, J.; Lou, H.; Jin, W.; Yang, L.; Pan, X.; Wang, J.; Shen, Y.; et al. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol. Biol. Evol. 2011, 28, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, H.; Liu, G.; Zhao, F.; Kijas, J.W.; Ma, Y.; Lu, J.; Zhang, L.; Cao, J.; Wu, M.; et al. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 2016, 6, 26770. [Google Scholar] [CrossRef]
- Guo, Y.-B.; He, Y.-X.; Cui, C.-Y.; Ou, Z.; Bai, M.; Duo, J.; De, J.; Bian, B.; Yi, P.; Bai, C.; et al. GCH1 plays a role in the high-altitude adaptation of Tibetans. Zool. Res. 2017, 38, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gou, W.; Wang, X.; Zhang, Y.; Ma, J.; Zhang, H.; Zhang, Y.; Zhang, H. Genome Resequencing Identifies Unique Adaptations of Tibetan Chickens to Hypoxia and High-Dose Ultraviolet Radiation in High-Altitude Environments. Genome Biol. Evol. 2016, 8, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Li, C.; Li, Z.; Chen, W.; Zeng, Y. Genetic Diversities and Differentially Selected Regions Between Shandong Indigenous Pig Breeds and Western Pig Breeds. Front. Genet. 2019, 10, 1351. [Google Scholar] [CrossRef]
- Zhang, Z.; Jia, Y.; Almeida, P.; Mank, J.E.; van Tuinen, M.; Wang, Q.; Jiang, Z.; Chen, Y.; Zhan, K.; Hou, S.; et al. Whole-genome resequencing reveals signatures of selection and timing of duck domestication. Gigascience 2018, 7, giy027. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, T.; Schönenberger, M.J.; Walter, K.M.; Charles, K.N.; Faust, P.L.; Kovacs, W.J. Peroxisome-Deficiency and HIF-2α Signaling Are Negative Regulators of Ketohexokinase Expression. Front. Cell Dev. Biol. 2020, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.J.; Prieto-Lloret, J.; Becker, S.; Ward, J.P.; Aaronson, P.I. Hypoxic pulmonary vasoconstriction in the absence of pretone: Essential role for intracellular Ca2+ release. J. Physiol. 2013, 591, 4473–4498. [Google Scholar] [CrossRef] [PubMed]
- Calbet, J.A.; Boushel, R.; Robach, P.; Hellsten, Y.; Saltin, B.; Lundby, C. Chronic hypoxia increases arterial blood pressure and reduces adenosine and ATP induced vasodilatation in skeletal muscle in healthy humans. Acta Physiol. 2014, 211, 574–584. [Google Scholar] [CrossRef]
- Crutzen, R.; Virreira, M.; Markadieu, N.; Shlyonsky, V.; Sener, A.; Malaisse, W.J.; Beauwens, R.; Boom, A.; Golstein, P.E. Anoctamin 1 (Ano1) is required for glucose-induced membrane potential oscillations and insulin secretion by murine β-cells. Pflug. Arch. 2016, 468, 573–591. [Google Scholar] [CrossRef]
- Edlund, A.; Esguerra, J.L.; Wendt, A.; Flodström-Tullberg, M.; Eliasson, L. CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med. 2014, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Neary, M.T.; Neary, J.M.; Lund, G.K.; Holt, T.N.; Garry, F.B.; Mohun, T.J.; Breckenridge, R.A. Myosin heavy chain 15 is associated with bovine pulmonary arterial pressure. Pulm. Circ. 2014, 4, 496–503. [Google Scholar] [CrossRef]
- Bai, X.; Lenhart, K.C.; Bird, K.E.; Suen, A.A.; Rojas, M.; Kakoki, M.; Li, F.; Smithies, O.; Mack, C.P.; Taylor, J.M. The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nat. Commun. 2013, 4, 2910. [Google Scholar] [CrossRef] [PubMed]
- Carney, E.F. Hypertension: Role of ARHGAP42 in hypertension. Nat. Rev. Nephrol. 2017, 13, 134. [Google Scholar] [CrossRef]
- Ayalew, W.; Chu, M.; Liang, C.; Wu, X.; Yan, P. Adaptation Mechanisms of Yak (Bos grunniens) to High-Altitude Environmental Stress. Animals 2021, 11, 2344. [Google Scholar] [CrossRef]
- Ma, W.; Li, X.; Shen, J.; Du, Y.; Xu, K.; Jiang, Y. Transcriptomic analysis reveals Apis mellifera adaptations to high temperature and high humidity. Ecotoxicol. Environ. Saf. 2019, 184, 109599. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, W.R.; Lv, F.H.; He, S.G.; Tian, S.L.; Peng, W.F.; Sun, Y.W.; Zhao, Y.X.; Tu, X.L.; Zhang, M.; et al. Whole-Genome Sequencing of Native Sheep Provides Insights into Rapid Adaptations to Extreme Environments. Mol. Biol. Evol. 2016, 33, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, E.A.; Timiras, P.S. Biological effects of high altitude as related to increased solar radiation, temperature fluctuations and reduced partial pressure of oxygen. Prog. Biometeorol. 1974, 1, 295–328,646–662. [Google Scholar]
- Miao, Y.W.; Peng, M.S.; Wu, G.S.; Ouyang, Y.N.; Yang, Z.Y.; Yu, N.; Liang, J.P.; Pianchou, G.; Beja-Pereira, A.; Mitra, B.; et al. Chicken domestication: An updated perspective based on mitochondrial genomes. Heredity 2013, 110, 277–282. [Google Scholar] [CrossRef]
- Tixier-Boichard, M.; Bed’hom, B.; Rognon, X. Chicken domestication: From archeology to genomics. Comptes Rendus. Biol. 2011, 334, 197–204. [Google Scholar] [CrossRef]
- Ginerete, R.P.; Mascio, G.; Liberatore, F.; Bucci, D.; Antenucci, N.; Di Pietro, P.; Cannella, M.; Imbriglio, T.; Notartomaso, S.; Nicoletti, F.; et al. Repeated episodes of transient reduction of oxygen exposure simulating aircraft cabin conditions enhance resilience to stress in mice. Eur. J. Neurosci. 2021, 54, 7109–7124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, D.; Zhang, Y.; Li, J.; Ma, S.; Zhang, J.; Xiong, Y.; Wang, W.; Li, N.; Xia, L. Knockdown of lncRNA BDNF-AS suppresses neuronal cell apoptosis via downregulating miR-130b-5p target gene PRDM5 in acute spinal cord injury. RNA Biol. 2018, 15, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.D.; Yang, C.P.; Wang, M.S.; Dong, K.Z.; Yan, D.W.; Hao, Z.Q.; Fan, S.Q.; Chu, S.Z.; Shen, Q.S.; Jiang, L.P.; et al. Convergent genomic signatures of high-altitude adaptation among domestic mammals. Natl. Sci. Rev. 2020, 7, 952–963. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Li, X.; Guo, Q.; Huang, L.; Bai, H.; Chang, G. Genome Sequencing Reveals the Adaptation of Chickens to High Altitudes in Different Regions. Animals 2025, 15, 265. https://doi.org/10.3390/ani15020265
Hu Y, Li X, Guo Q, Huang L, Bai H, Chang G. Genome Sequencing Reveals the Adaptation of Chickens to High Altitudes in Different Regions. Animals. 2025; 15(2):265. https://doi.org/10.3390/ani15020265
Chicago/Turabian StyleHu, Yizhou, Xing Li, Qixin Guo, Lan Huang, Hao Bai, and Guobin Chang. 2025. "Genome Sequencing Reveals the Adaptation of Chickens to High Altitudes in Different Regions" Animals 15, no. 2: 265. https://doi.org/10.3390/ani15020265
APA StyleHu, Y., Li, X., Guo, Q., Huang, L., Bai, H., & Chang, G. (2025). Genome Sequencing Reveals the Adaptation of Chickens to High Altitudes in Different Regions. Animals, 15(2), 265. https://doi.org/10.3390/ani15020265