
Successful Traceability of Wildlife Samples Contributes to Wildlife Conservation: A Case Study of Tracing the Snub-Nosed Monkey (Rhinopithecus spp.)

 ,
,
Simple Summary
Abstract
1. Introduction
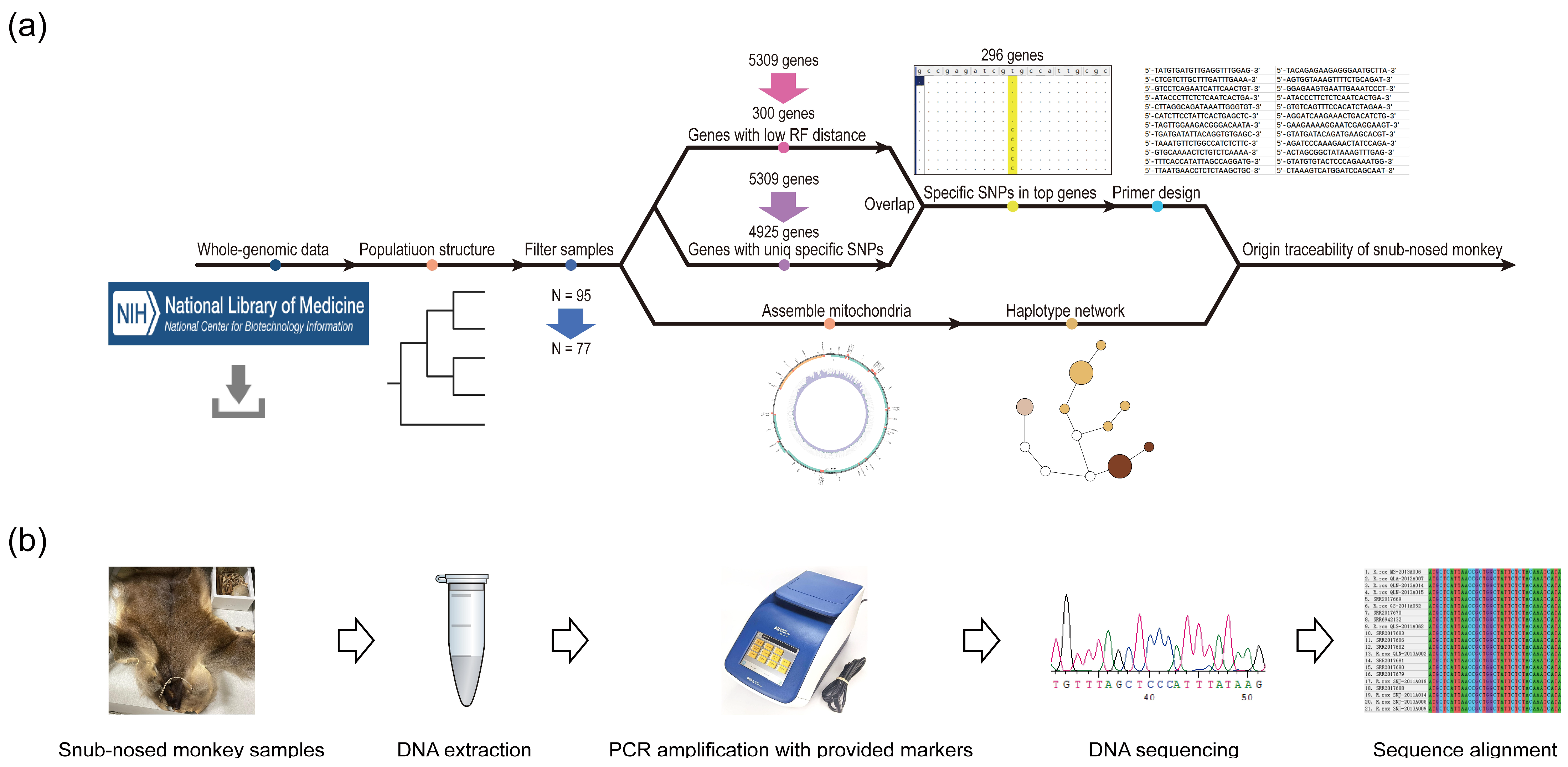
2. Materials and Methods
2.1. Single Nucleotide Polymorphism (SNP) Genotyping
2.2. Filtering of Potentially Related and Mixed Individuals
2.3. Mitochondrial Genome Assembly and Network Construction
2.4. Population Structure and Admixture Analyses
2.5. Scanning of Whole-Genome Specific Loci and Primer Design
2.6. DNA Extraction and PCR Validation
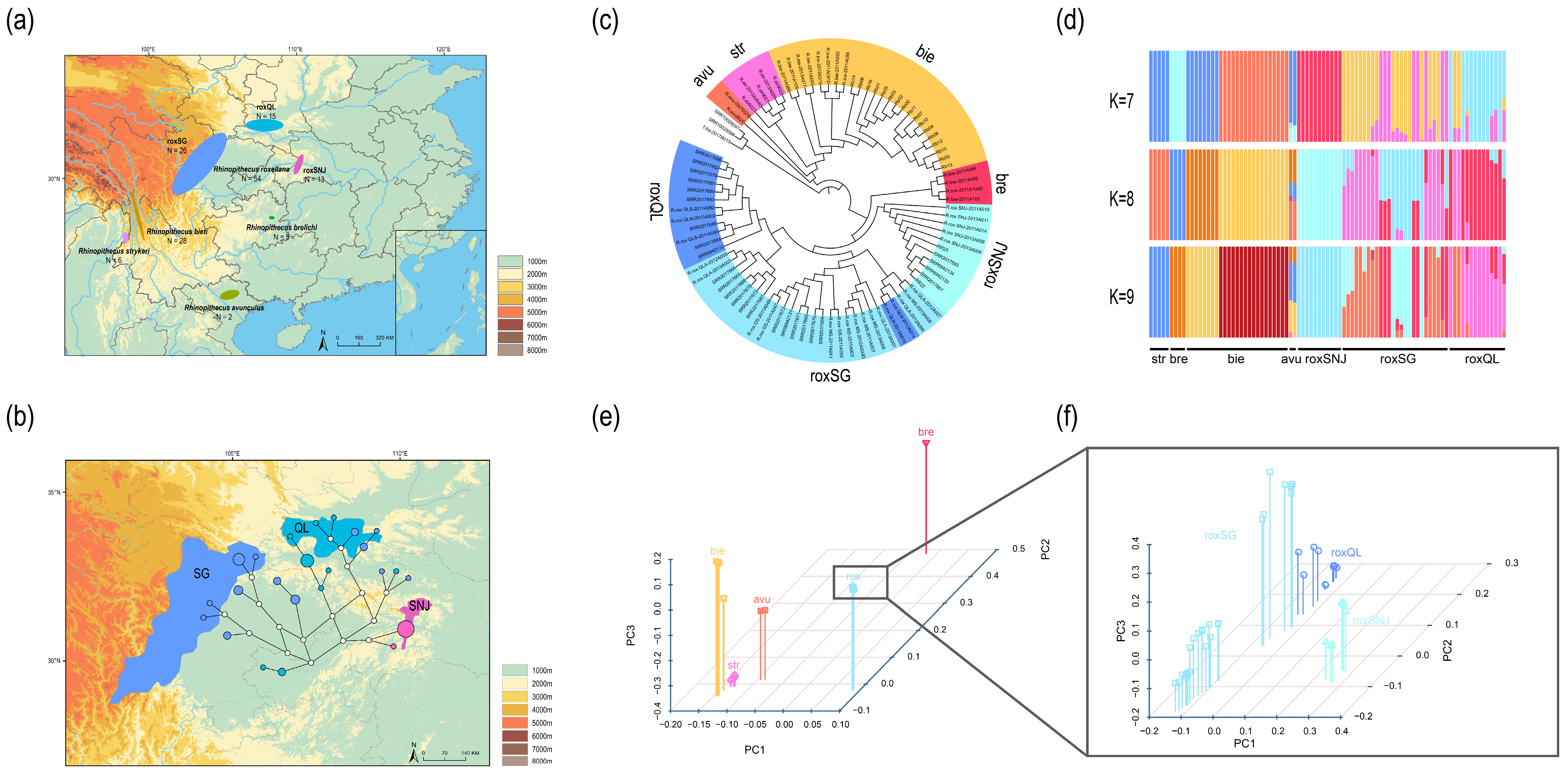
3. Results
3.1. Variant Calling and Filtering of Samples Derived from Potentially Mixed Individuals
3.2. Mitochondrial Genome Assembly and Network Construction
3.3. Scanning of Specific Loci of nDNA
3.4. Traceability Analysis and Primer Validation
4. Discussion
4.1. Mitochondrial DNA Provides Low Resolution for Population Level Traceability of R. roxellana
4.2. Applying WGS Data for Traceability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jablonski, N.G. The response of catarrhine primates to pleistocene environmental fluctuations in east Asia. Primates 1998, 39, 29–37. [Google Scholar] [CrossRef]
- Li, B.G.; Pan, R.L.; Oxnard, C.E. Extinction of snub-nosed monkeys in China during the past 400 years. Int. J. Primatol. 2002, 23, 1227–1244. [Google Scholar] [CrossRef]
- Quan, G.Q.; Xie, J.Y. Research on the Golden Monkey; Science and Education Publishing House: Beijing, China, 2002; pp. 79–92. [Google Scholar]
- Kuang, W.; Hu, J.; Wu, H.; Fen, X.; Dai, Q.; Fu, Q.; Xiao, W.; Frantz, L.; Roos, C.; Nadler, T.; et al. Genetic diversity, inbreeding level, and genetic load in endangered snub-nosed monkeys (Rhinopithecus). Front. Genet. 2020, 11, 615926. [Google Scholar] [CrossRef] [PubMed]
- Kuderna, L.F.K.; Gao, H.; Janiak, M.C.; Kuhlwilm, M.; Orkin, J.D.; Bataillon, T.; Manu, S.; Valenzuela, A.; Bergman, J.; Rousselle, M.; et al. A global catalog of whole-genome diversity from 233 primate species. Science 2023, 380, 906–913. [Google Scholar] [CrossRef]
- Zhao, X.; Ren, B.; Garber, P.A.; Li, X.; Li, M. Impacts of human activity and climate change on the distribution of snub-nosed monkeys in china during the past 2000 years. Divers. Distrib. 2018, 24, 92–102. [Google Scholar] [CrossRef]
- Tinsman, J.C.; Gruppi, C.; Bossu, C.M.; Prigge, T.; Harrigan, R.J.; Zaunbrecher, V.; Koepfli, K.; Lebreton, M.; Njabo, K.; Wenda, C.; et al. Genomic analyses reveal poaching hotspots and illegal trade in pangolins from Africa to Asia. Science 2023, 382, 1282–1286. [Google Scholar] [CrossRef]
- Salum, J.; Eustace, A.; Malata, P.F.; Mbangwa, O.F. Wildlife crime promoted by weak governance. Afr. J. Ecol. 2018, 56, 101–108. [Google Scholar] [CrossRef]
- Harper, C.K. Poaching forensics: Animal victims in the courtroom. Annu. Rev. Anim. Biosci. 2023, 11, 269–286. [Google Scholar] [CrossRef]
- Ogden, R. Unlocking the potential of genomic technologies for wildlife forensics. Mol. Ecol. Resour. 2011, 11, 109–116. [Google Scholar] [CrossRef]
- Morgan, R.M.; Wiltshire, P.; Parker, A.; Bull, P.A. The role of forensic geoscience in wildlife crime detection. Forensic Sci. Int. 2006, 162, 152–162. [Google Scholar] [CrossRef]
- Johnson, R.N.; Wilson-Wilde, L.; Linacre, A. Current and future directions of DNA in wildlife forensic science. Forensic Sci. Int. Genet. 2014, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Georgiadis, N.J.; Hondo, T.; Roca, A.L. Triangulating the provenance of African elephants using mitochondrial DNA. Evol. Appl. 2013, 6, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Nagata, J.; Aramilev, V.V.; Belozor, A.; Higashi, S.; Mccullough, D.R. Species and sex identification from faecal samples of sympatric carnivores, amur leopard and Siberian tiger, in the Russian far east. Conserv. Genet. 2006, 7, 799–802. [Google Scholar] [CrossRef]
- Wasser, S.K.; Shedlock, A.M.; Comstock, K.; Ostrander, E.A.; Mutayoba, B.; Stephens, M. Assigning african elephant dna to geographic region of origin: Applications to the ivory trade. Proc. Natl. Acad. Sci. USA 2004, 101, 14847–14852. [Google Scholar] [CrossRef]
- Pfeiffer, I.; Völkel, I.; Täubert, H.; Brenig, B. Forensic DNA-typing of dog hair: DNA-extraction and PCR amplification. Forensic Sci. Int. 2004, 141, 149–151. [Google Scholar] [CrossRef]
- Xie, L.; Deng, Y.; Shao, X.; Hu, P.; Zhao, D.; Li, C.; Ba, H. Design of a universal primer pair for the identification of deer species. Conserv. Genet. Resour. 2021, 13, 9–12. [Google Scholar] [CrossRef]
- Oliveira, R.; Castro, D.; Godinho, R.; Luikart, G.; Alves, P.C. Species identification using a small nuclear gene fragment: Application to sympatric wild carnivores from south-western Europe. Conserv. Genet. 2010, 11, 1023–1032. [Google Scholar] [CrossRef]
- Paneto, G.G.; Martins, J.A.; Longo, L.V.G.; Pereira, G.A.; Freschi, A.; Alvarenga, V.L.S.; Chen, B.; Oliveira, R.N.; Hirata, M.H.; Cicarelli, R.M.B. Heteroplasmy in hair: Differences among hair and blood from the same individuals are still a matter of debate. Forensic Sci. Int. 2007, 173, 117–121. [Google Scholar] [CrossRef]
- Kim, J.H.; Antunes, A.; Luo, S.J.; Menninger, J.; Nash, W.G.; O’Brien, S.J.; Johnson, W.E. Evolutionary analysis of a large mtdna translocation (numt) into the nuclear genome of the panthera genus species. Gene 2006, 366, 292–302. [Google Scholar] [CrossRef]
- Lopez, J.V.; Yuhki, N.; Masuda, R.; Modi, W.; Obrien, S.J. Numt, a recent transfer and tandem amplification of mitochondrial-DNA to the nuclear genome of the domestic cat. J. Mol. Evol. 1994, 39, 544. [Google Scholar] [CrossRef]
- Spinks, P.Q.; Thomson, R.C.; Zhang, Y.; Che, J.; Wu, Y.; Shaffer, H.B. Species boundaries and phylogenetic relationships in the critically endangered Asian box turtle genus cuora. Mol. Phylogenet. Evol. 2012, 63, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Alacs, E.A.; Georges, A.; Fitzsimmons, N.N.; Robertson, J. DNA detective: A review of molecular approaches to wildlife forensics. Forensic Sci. Med. Pathol. 2010, 6, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.; Ming, C.; Li, H.; Wu, H.; Frantz, L.; Roos, C.; Zhang, Y.; Zhang, C.; Jia, T.; Yang, J.; et al. The origin and population history of the endangered golden snub-nosed monkey (Rhinopithecus roxellana). Mol. Biol. Evol. 2019, 36, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, G.; Ruan, J.; Chen, Y.; Yang, C.; Cao, X.; Wu, H.; Liu, Y.; Du, Z.; Wang, X.; et al. Genomic analysis of snub-nosed monkeys (Rhinopithecus) identifies genes and processes related to high-altitude adaptation. Nat. Genet. 2016, 48, 947. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.M.; Meng, X.H.; Liu, Z.J.; Chang, J.; Wang, B.S.; Li, M.Z.; Orozco-Ter Wengel, P.; Tian, S.L.; Wen, C.L.; Wang, Z.M.; et al. Population genomics reveals low genetic diversity and adaptation to hypoxia in snub-nosed monkeys. Mol. Biol. Evol. 2016, 33, 2670–2681. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, L.; Yan, Z.; Ren, Z.; Han, F.; Tan, X.; Xiang, Z.; Dong, F.; Yang, Z.; Liu, G.; et al. Genomic mechanisms of physiological and morphological adaptations of limestone langurs to karst habitats. Mol. Biol. Evol. 2019, 37, 952–968. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Mckenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; Depristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and vcftools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Tung, N.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpeff: Snps in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite—Fast d-statistics and related admixture evidence from vcf files. Mol. Ecol. Resour. 2021, 21, 584–595. [Google Scholar] [CrossRef]
- Iyengar, A. Forensic dna analysis for animal protection and biodiversity conservation: A review. J. Nat. Conserv. 2014, 22, 195–205. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. Mitoz: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids. Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. Novoplasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids. Res. 2017, 45, e18. [Google Scholar]
- Chi, L.; Zhang, X.; Xue, Y.; Chen, H. Fasthan: A fast and scalable program for constructing haplotype network for large-sample sequences. Mol. Ecol. Resour. 2023, 1–5. [Google Scholar] [CrossRef]
- Murias, D.S.A.; Cabezas, M.P.; Tavares, A.I.; Xavier, R.; Branco, M. Tcsbu: A tool to extend tcs network layout and visualization. Bioinformatics 2016, 32, 627–628. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. Raxml-ng: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (itol) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids. Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Leslie, S.; Winney, B.; Hellenthal, G.; Davison, D.; Boumertit, A.; Day, T.; Hutnik, K.; Royrvik, E.C.; Cunliffe, B.; Lawson, D.J.; et al. The fine-scale genetic structure of the british population. Nature 2015, 519, 309. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.J.; Hellenthal, G.; Myers, S.; Falush, D. Inference of population structure using dense haplotype data. PLoS Genet. 2012, 8, e1002453. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.F.; Foulds, L.R. Comparison of phylogenetic trees. Math. Biosci. 1981, 53, 131–147. [Google Scholar] [CrossRef]
- Ginestet, C. Ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. A-Stat. Soc. 2011, 174, 245. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. Upsetr: An r package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. Mega11 molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; Mccarthy, S.A.; Davies, R.M.; et al. Twelve years of samtools and bcftools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids. Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Winnepenninckx, B.; Backeljau, T.; De Wachter, R. Extraction of high molecular weight DNA from Molluscs. Trends Genet. TIG 1993, 9, 407. [Google Scholar]
- Wang, L.; Wu, J.; Liu, X.; Di, D.; Liang, Y.; Feng, Y.; Zhang, S.; Li, B.; Qi, X. A high-quality genome assembly for the endangered golden snub-nosed monkey (Rhinopithecus roxellana). Gigascience 2019, 8, giz098. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Song, S.; Li, C.; Zhang, J. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature 2022, 606, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Kanthaswamy, S. Wildlife forensic genetics-biological evidence, DNA markers, analytical approaches, and challenges. Anim. Genet. 2024, 55, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Akutsu, T.; Watanabe, K.; Ogawa, Y.; Imaizumi, K. A vertebrate-specific qpcr assay as an endogenous internal control for robust species identification. Forensic Sci. Int. Genet. 2022, 56, 102628. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Intraspecific gene genealogies: Trees grafting into networks. Trends Ecol. Evol. 2001, 16, 37–45. [Google Scholar] [CrossRef]
- Harl, J.; Himmel, T.; Valkiunas, G.; Ilgunas, M.; Bakonyi, T.; Weissenboeck, H. Geographic and host distribution of haemosporidian parasite lineages from birds of the family turdidae. Malar. J. 2020, 19, 335. [Google Scholar] [CrossRef]
- Fuhrmann, N.; Kaiser, T.S. The importance of DNA barcode choice in biogeographic analyses a case study on marine midges of the genus clunio. Genome 2021, 64, 242–252. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; Wilson, R.K.; et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 2007, 316, 222–234. [Google Scholar] [CrossRef]
- King, J.L.; Jukes, T.H. Non-darwinian evolution. Science 1969, 164, 788–798. [Google Scholar] [CrossRef]
- Liu, J.; Milne, R.I.; Moller, M.; Zhu, G.; Ye, L.; Luo, Y.; Yang, J.; Wambulwa, M.C.; Wang, C.; Li, D.; et al. Integrating a comprehensive DNA barcode reference library with a global map of yews (Taxus L.) For forensic identification. Mol. Ecol. Resour. 2018, 18, 1115–1131. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Z.; Zhang, Y.; Frantz, L.; Roos, C.; Irwin, D.M.; Zhang, C.; Liu, X.; Wu, D.; Huang, S.; et al. Hybrid origin of a primate, the gray snub-nosed monkey. Science 2023, 380, 926. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.; Oliveira, L.R.; Beux, Y.; Kessler, A.; Cardenas-Alayza, S.; Majluf, P.; Paez-Rosas, D.; Chaves, J.; Crespo, E.; Brownell, R.L.J.; et al. Genomic evidence for homoploid hybrid speciation in a marine mammal apex predator. Sci. Adv. 2023, 9, eadf6601. [Google Scholar] [CrossRef] [PubMed]
- Bowen, G.J.; Wassenaar, L.I.; Hobson, K.A. Global application of stable hydrogen and oxygen isotopes to wildlife forensics. Oecologia 2005, 143, 337–348. [Google Scholar] [CrossRef]
- Meier-Augenstein, W. From stable isotope ecology to forensic isotope ecology—Isotopes’ tales. Forensic Sci. Int. 2019, 300, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Picard, C.; Manicke, N.; Dembinski, G. Utilizing phormia regina as an environmental sensor for resource identification and biodiversity monitoring. Available online: https://scholarworks.indianapolis.iu.edu/items/fc6517d5-0ad3-4676-b496-88d30e88261a (accessed on 22 November 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Gene | Forward | Reverse | Amplicon Length |
|---|---|---|---|---|
| str | OCA2 | 5′-CTCGTCTTGCTTTGATTTGAAA-3′ | 5′-AGTGGTAAAGTTTTCTGCAGAT-3′ | 817 bp |
| bre | OCA2 | 5′-GTCCTCAGAATCATTCAACTGT-3′ | 5′-GGAGAAGTGAATTGAAATCCCT-3′ | 865 bp |
| bie | OCA2 | 5′-TTCCTCCATCCTCTAGTATTCC-3′ | 5′-ACAACAACAACAAAAGTAACCC-3′ | 972 bp |
| avu | MX2 | 5′-TAGTTGGAAGACGGGACAATA-3′ | 5′-GAAGAAAAGGAATCGAGGAAGT-3′ | 882 bp |
| rox | GART | 5′-CTGGGCATCTGAAAATAGACAT-3′ | 5′-GTGTGTGTGTATATAGGCCTTT-3′ | 878 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Shen, Y.; Teng, Y.; Wu, R.; Liu, S.; Zhao, J.; Hu, C.; Li, M.; Pan, H.; Qi, J. Successful Traceability of Wildlife Samples Contributes to Wildlife Conservation: A Case Study of Tracing the Snub-Nosed Monkey (Rhinopithecus spp.). Animals 2025, 15, 174. https://doi.org/10.3390/ani15020174
Wang X, Shen Y, Teng Y, Wu R, Liu S, Zhao J, Hu C, Li M, Pan H, Qi J. Successful Traceability of Wildlife Samples Contributes to Wildlife Conservation: A Case Study of Tracing the Snub-Nosed Monkey (Rhinopithecus spp.). Animals. 2025; 15(2):174. https://doi.org/10.3390/ani15020174
Chicago/Turabian StyleWang, Xibo, Ying Shen, Yang Teng, Ruifeng Wu, Shuhao Liu, Jilai Zhao, Can Hu, Ming Li, Huijuan Pan, and Jiwei Qi. 2025. "Successful Traceability of Wildlife Samples Contributes to Wildlife Conservation: A Case Study of Tracing the Snub-Nosed Monkey (Rhinopithecus spp.)" Animals 15, no. 2: 174. https://doi.org/10.3390/ani15020174
APA StyleWang, X., Shen, Y., Teng, Y., Wu, R., Liu, S., Zhao, J., Hu, C., Li, M., Pan, H., & Qi, J. (2025). Successful Traceability of Wildlife Samples Contributes to Wildlife Conservation: A Case Study of Tracing the Snub-Nosed Monkey (Rhinopithecus spp.). Animals, 15(2), 174. https://doi.org/10.3390/ani15020174