Uncovering Molecular Mechanisms of Feed Efficiency in Pigs Through Multi-Omics Analysis of the Jejunum

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Jejunal Samples
2.2. Generation of RNA-Seq Libraries
2.3. Analysis of RNA-Seq Data
2.4. Generation of ATAC-Seq Libraries
2.5. Analysis of ATAC-Seq Data
2.6. Functional Annotation of Genes and Peaks
2.7. Construction of Gene Regulatory Network
2.8. Validation of Transcription Factors and Genes in Gene Regulatory Network
3. Results
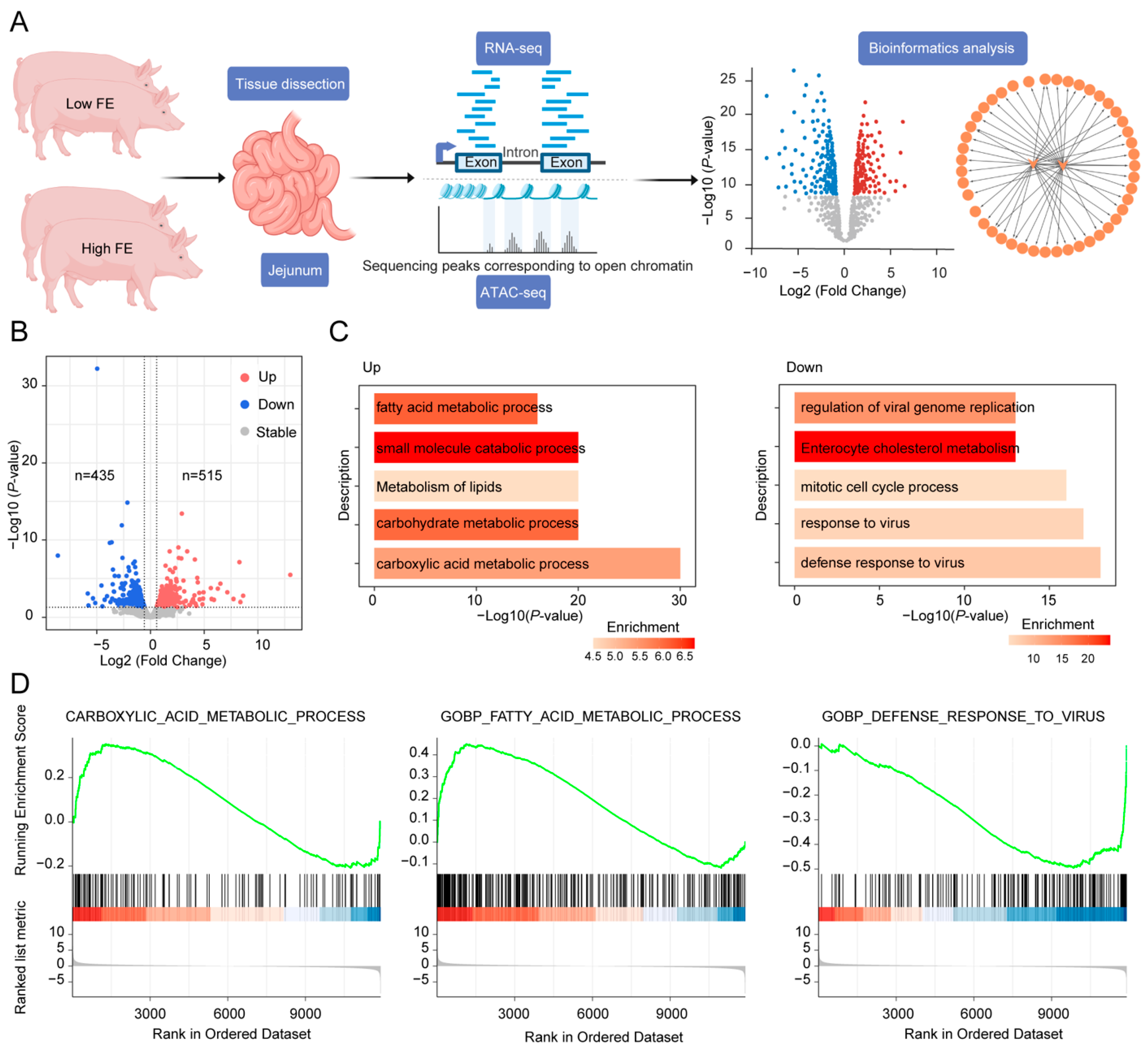
3.1. Identification of DEGs in the Jejunum Between Pigs with High and Low FE
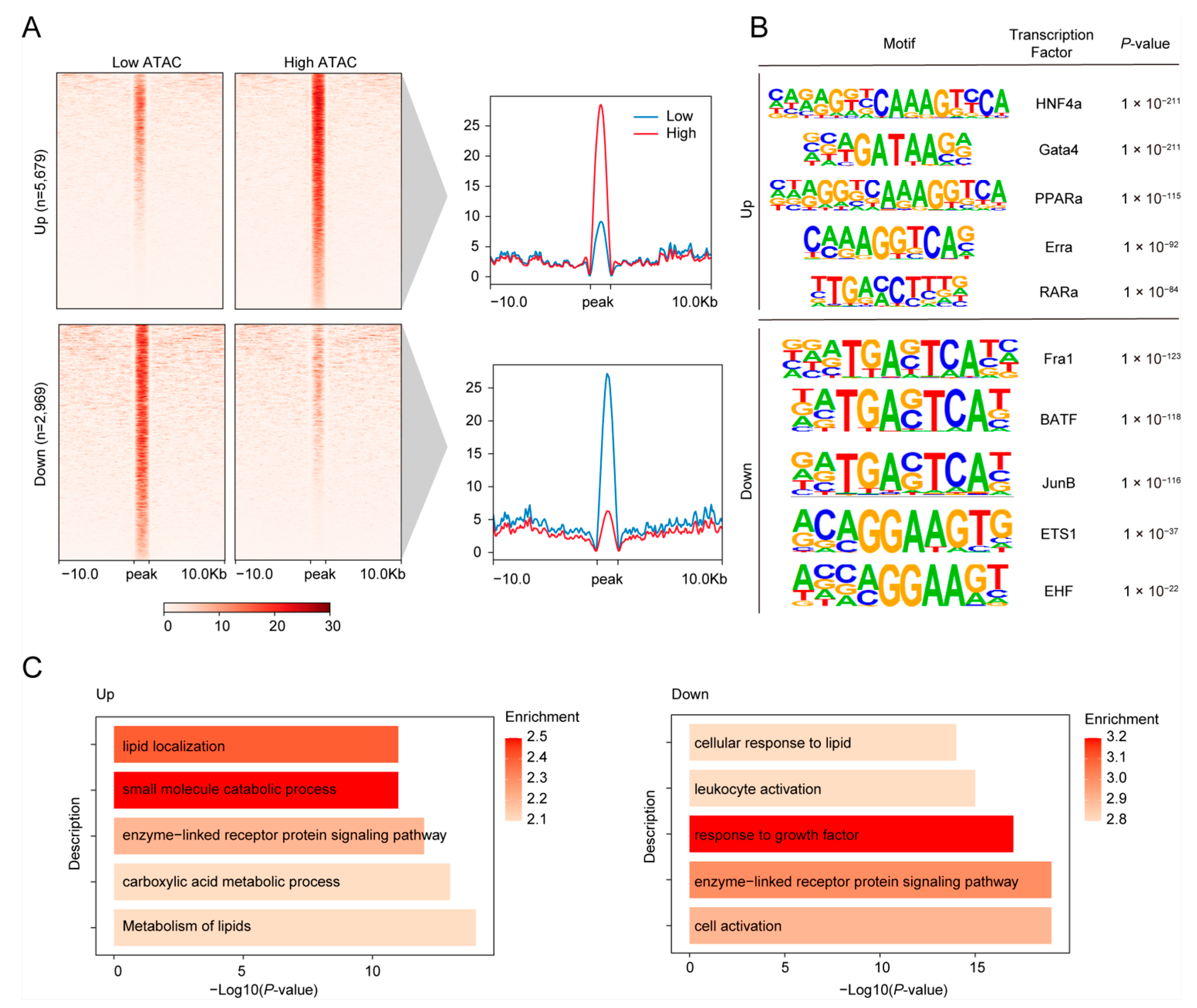
3.2. Identification and Characterization of Candidate CREs in the Jejunum of High and Low FE Pigs
3.3. Differential Analysis of Chromatin Accessible cCREs Between Low and High FE Pigs
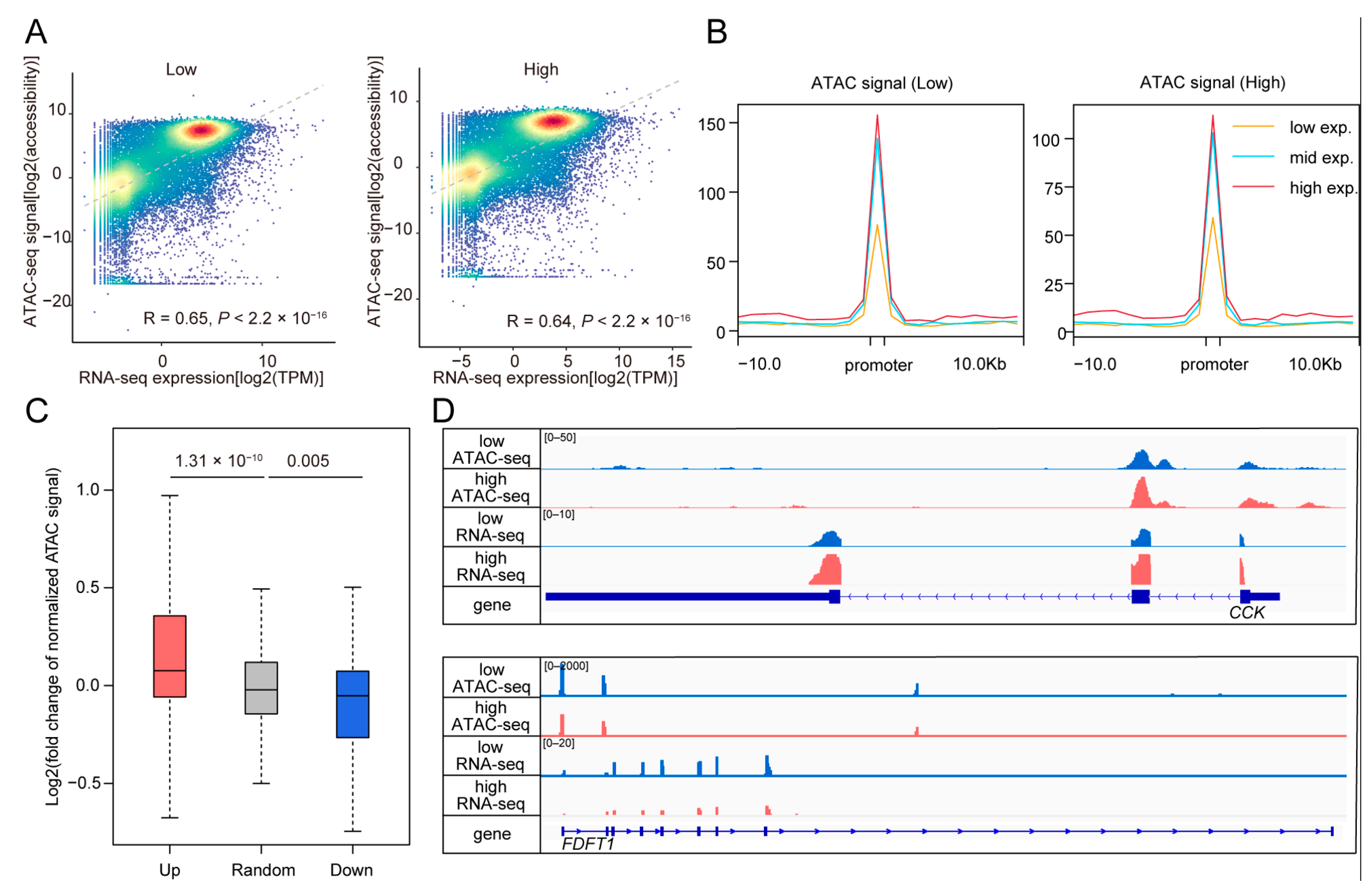
3.4. Comparison of Gene Expression and Chromatin Accessibility Revealing High Consistency Between Two Modalities
3.5. Integrating DEGs and DARs to Construct a FE-Related Regulatory Gene Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patience, J.F.; Rossoni-Serao, M.C.; Gutierrez, N.A. A review of feed efficiency in swine: Biology and application. J. Anim. Sci. Biotechnol. 2015, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, T.; Gilbert, H. An approach to achieve overall farm feed efficiency in pig production: Environmental evaluation through individual life cycle assessment. Int. J. Life Cycle Assess. 2021, 26, 455–469. [Google Scholar] [CrossRef]
- Kumar, P.; Abubakar, A.A.; Verma, A.K.; Umaraw, P.; Adewale Ahmed, M.; Mehta, N.; Nizam Hayat, M.; Kaka, U.; Sazili, A.Q. New insights in improving sustainability in meat production: Opportunities and challenges. Crit. Rev. Food Sci. Nutr. 2023, 63, 11830–11858. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Kachungwa Lugata, J.; Ortega, A. Gut Health and Influencing Factors in Pigs. Animals 2023, 13, 1350. [Google Scholar] [CrossRef] [PubMed]
- Wijtten, P.J.; van der Meulen, J.; Verstegen, M.W. Intestinal barrier function and absorption in pigs after weaning: A review. Br. J. Nutr. 2011, 105, 967–981. [Google Scholar] [CrossRef]
- Kvidera, S.K.; Mayorga, E.J.; McCarthy, C.S.; Horst, E.A.; Abeyta, M.A.; Baumgard, L.H. Effects of supplemental citrulline on thermal and intestinal morphology parameters during heat stress and feed restriction in growing pigs. J. Anim. Sci. 2024, 102, skae120. [Google Scholar] [CrossRef]
- Duarte, M.E.; Sparks, C.; Kim, S.W. Modulation of jejunal mucosa-associated microbiota in relation to intestinal health and nutrient digestibility in pigs by supplementation of β-glucanase to corn–soybean meal-based diets with xylanase. J. Anim. Sci. 2021, 99, skab190. [Google Scholar] [CrossRef]
- Metzler-Zebeli, B.U.; Lawlor, P.G.; Magowan, E.; McCormack, U.M.; Curiao, T.; Hollmann, M.; Ertl, R.; Aschenbach, J.R.; Zebeli, Q. Finishing pigs that are divergent in feed efficiency show small differences in intestinal functionality and structure. PLoS ONE 2017, 12, e0174917. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ding, S.; Azad, M.A.K.; Song, B.; Kong, X. Small Intestinal Digestive Functions and Feed Efficiency Differ in Different Pig Breeds. Animals 2023, 13, 1172. [Google Scholar] [CrossRef] [PubMed]
- Reszka, P.; Dunislawska, A.; Slawinska, A.; Siwek, M.; Kapelanski, W.; Bogucka, J. Influence of the effective microorganisms (EM) on performance, intestinal morphology and gene expression in the jejunal mucosa of pigs fed different diets. J. Anim. Physiol. Anim. Nutr. 2020, 104, 1444–1453. [Google Scholar] [CrossRef]
- Xiang, Y.; Sun, J.; Ma, G.; Dai, X.; Meng, Y.; Fu, C.; Zhang, Y.; Zhao, Q.; Li, J.; Zhang, S.; et al. Integrating Multi-Omics Data to Identify Key Functional Variants Affecting Feed Efficiency in Large White Boars. Genes 2024, 15, 980. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kahari, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranasic, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids. Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Gondret, F.; Vincent, A.; Houee-Bigot, M.; Siegel, A.; Lagarrigue, S.; Causeur, D.; Gilbert, H.; Louveau, I. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genom. 2017, 18, 244. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, P.; Do, D.N.; Colombo, S.M.; Rathgeber, B.; Miar, Y. Application of Genetic, Genomic and Biological Pathways in Improvement of Swine Feed Efficiency. Front. Genet. 2022, 13, 903733. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Yang, M.; Wang, X.; Cai, G.; Ding, R.; Zhuang, Z.; Zhou, S.; Tan, S.; Ruan, D.; Wu, J.; et al. Multi-omic characterization of allele-specific regulatory variation in hybrid pigs. Nat. Commun. 2024, 15, 5587. [Google Scholar] [CrossRef]
- Bai, J.; Lin, Y.; Zhang, J.; Chen, Z.; Wang, Y.; Li, M.; Li, J. Profiling of Chromatin Accessibility in Pigs across Multiple Tissues and Developmental Stages. Int. J. Mol. Sci. 2023, 24, 1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hocker, J.D.; Miller, M.; Hou, X.; Chiou, J.; Poirion, O.B.; Qiu, Y.; Li, Y.E.; Gaulton, K.J.; Wang, A.; et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 2021, 184, 5985–6001.e5919. [Google Scholar] [CrossRef]
- Vemuri, K.; Radi, S.H.; Sladek, F.M.; Verzi, M.P. Multiple roles and regulatory mechanisms of the transcription factor HNF4 in the intestine. Front. Endocrinol. 2023, 14, 1232569. [Google Scholar] [CrossRef] [PubMed]
- Girard, R.; Tremblay, S.; Noll, C.; St-Jean, S.; Jones, C.; Gelinas, Y.; Maloum-Rami, F.; Perreault, N.; Laplante, M.; Carpentier, A.C.; et al. The transcription factor hepatocyte nuclear factor 4A acts in the intestine to promote white adipose tissue energy storage. Nat. Commun. 2022, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Patankar, J.V.; Obrowsky, S.; Doddapattar, P.; Hoefler, G.; Battle, M.; Levak-Frank, S.; Kratky, D. Intestinal GATA4 deficiency protects from diet-induced hepatic steatosis. J. Hepatol. 2012, 57, 1061–1068. [Google Scholar] [CrossRef]
- Patankar, J.V.; Chandak, P.G.; Obrowsky, S.; Pfeifer, T.; Diwoky, C.; Uellen, A.; Sattler, W.; Stollberger, R.; Hoefler, G.; Heinemann, A.; et al. Loss of intestinal GATA4 prevents diet-induced obesity and promotes insulin sensitivity in mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E478–E488. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, R.; Zheng, M.; Feng, F.; Liu, D.; Guo, Y.; Zhao, G.; Wen, J. New insights into the associations among feed efficiency, metabolizable efficiency traits and related QTL regions in broiler chickens. J. Anim. Sci. Biotechnol. 2020, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Reehorst, C.M.; Nightingale, R.; Luk, I.Y.; Jenkins, L.; Koentgen, F.; Williams, D.S.; Darido, C.; Tan, F.; Anderton, H.; Chopin, M. EHF is essential for epidermal and colonic epithelial homeostasis, and suppresses Apc-initiated colonic tumorigenesis. Development 2021, 148, dev199542. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Li, X.; Luo, W.; Xu, Z.; Ji, C.; Zhang, Y.; Nie, Q.; Zhang, D.; Zhang, X. Feed conversion ratio, residual feed intake and cholecystokinin type A receptor gene polymorphisms are associated with feed intake and average daily gain in a Chinese local chicken population. J. Anim. Sci. Biotechnol. 2018, 9, 50. [Google Scholar] [CrossRef]
- Dong, X.; Zhu, Y.; Wang, S.; Luo, Y.; Lu, S.; Nan, F.; Sun, G.; Sun, X. Bavachinin inhibits cholesterol synthesis enzyme FDFT1 expression via AKT/mTOR/SREBP-2 pathway. Int. Immunopharmacol. 2020, 88, 106865. [Google Scholar] [CrossRef]
- Stine, R.R.; Sakers, A.P.; TeSlaa, T.; Kissig, M.; Stine, Z.E.; Kwon, C.W.; Cheng, L.; Lim, H.W.; Kaestner, K.H.; Rabinowitz, J.D.; et al. PRDM16 Maintains Homeostasis of the Intestinal Epithelium by Controlling Region-Specific Metabolism. Cell Stem Cell 2019, 25, 830–845.e838. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, R.; Wang, J.; Zhang, Y.; Xing, S.; Zheng, M.; Cui, H.; Li, Q.; Li, P.; Cui, X.; et al. Exploring Genomic Variants Related to Residual Feed Intake in Local and Commercial Chickens by Whole Genomic Resequencing. Genes 2018, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ordovas, J.M.; Gao, G.; Province, M.; Straka, R.J.; Tsai, M.Y.; Lai, C.-Q.; Zhang, K.; Borecki, I.; Hixson, J.E. The SCARB1 gene is associated with lipid response to dietary and pharmacological interventions. J. Hum. Genet. 2008, 53, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Strathe, A.B.; Jensen, J.; Mark, T.; Kadarmideen, H.N. Genetic parameters for different measures of feed efficiency and related traits in boars of three pig breeds. J. Anim. Sci. 2013, 91, 4069–4079. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fang, S.; Yang, H.; Chen, C. Identification of the relationship between the gut microbiome and feed efficiency in a commercial pig cohort. J. Anim. Sci. 2021, 99, skab045. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.M.; Calo, J.; Soengas, J.L. The gut-brain axis in vertebrates: Implications for food intake regulation. J. Exp. Biol. 2021, 224, jeb231571. [Google Scholar] [CrossRef]
- Xie, Y.; Zhou, G.; Wang, C.; Xu, X.; Li, C. Specific Microbiota Dynamically Regulate the Bidirectional Gut-Brain Axis Communications in Mice Fed Meat Protein Diets. J. Agric. Food Chem. 2019, 67, 1003–1017. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Xiang, Y.; Jian, Y.; Zhao, Q.; Sun, J.; Huang, Y.; Xu, J.; Qi, X.; Li, J.; Zheng, Z.; et al. Uncovering Molecular Mechanisms of Feed Efficiency in Pigs Through Multi-Omics Analysis of the Jejunum. Animals 2025, 15, 137. https://doi.org/10.3390/ani15020137
Zhang S, Xiang Y, Jian Y, Zhao Q, Sun J, Huang Y, Xu J, Qi X, Li J, Zheng Z, et al. Uncovering Molecular Mechanisms of Feed Efficiency in Pigs Through Multi-Omics Analysis of the Jejunum. Animals. 2025; 15(2):137. https://doi.org/10.3390/ani15020137
Chicago/Turabian StyleZhang, Saixian, Yue Xiang, Yaobang Jian, Qiulin Zhao, Jiahui Sun, Yi Huang, Jing Xu, Xiaolong Qi, Jingjin Li, Zhuqing Zheng, and et al. 2025. "Uncovering Molecular Mechanisms of Feed Efficiency in Pigs Through Multi-Omics Analysis of the Jejunum" Animals 15, no. 2: 137. https://doi.org/10.3390/ani15020137
APA StyleZhang, S., Xiang, Y., Jian, Y., Zhao, Q., Sun, J., Huang, Y., Xu, J., Qi, X., Li, J., Zheng, Z., Fu, L., Liu, Y., & Li, X. (2025). Uncovering Molecular Mechanisms of Feed Efficiency in Pigs Through Multi-Omics Analysis of the Jejunum. Animals, 15(2), 137. https://doi.org/10.3390/ani15020137