1. Introduction
Emerging and re-emerging RNA viruses continue to pose substantial threats to both human and animal populations, often originating in animal reservoirs and exhibiting zoonotic potential. Among these, influenza A virus (IAV) remains a prototypical example of a veterinary virus capable of cross-species transmission with pandemic consequences. Its segmented RNA genome, coupled with the high mutation rate and ability to undergo antigenic drift and shift, enables rapid adaptation to new hosts, including humans, pigs, birds, and other mammals [
1,
2]. These properties not only complicate vaccination and antiviral strategies but also underscore the need for coordinated surveillance systems to monitor circulating strains [
3]. The continuing emergence of highly pathogenic avian influenza (HPAI) strains in domestic and wild animals reflects this dynamic evolution and highlights the relevance of veterinary virology in global pandemic preparedness.
Similarly, flaviviruses such as Zika virus (ZIKV) and dengue virus (DENV) represent serious public and veterinary health challenges. These arthropod-borne viruses are widely distributed in tropical and subtropical regions and are known to cause significant morbidity and mortality across various species. The 2015–2016 ZIKV outbreak in the Americas, associated with congenital brain malformations, as well as the ongoing burden of DENV with an estimated 390 million annual infections, exemplify their capacity for rapid geographic expansion and severe clinical outcomes [
4,
5]. Importantly, the spread of these flaviviruses is facilitated by global environmental changes, vector expansion, and increased human–animal interface, necessitating integrative One Health strategies to understand and control their epidemiology.
A common feature of these RNA viruses is their interaction with host cell death pathways, particularly apoptosis and necroptosis. While apoptosis traditionally acts as a host defense mechanism to limit viral replication by eliminating infected cells, certain viruses manipulate this pathway to their advantage [
6]. Recent studies have shown that viruses such as IAV, ZIKV, and DENV induce necroptotic signaling, potentially contributing to tissue damage and immune modulation [
7,
8]. Conversely, apoptotic processes—when well-regulated—can facilitate containment of infection through the formation of apoptotic bodies that encapsulate viral particles, preventing their release and promoting clearance by phagocytes [
6,
9]. Thus, understanding how veterinary viruses interface with host immunological responses, including programmed cell death, is critical to elucidating their pathogenesis.
In response to these complex host–virus interactions and the limitations of current antiviral therapies, there has been increasing interest in the discovery of broad-spectrum antiviral compounds from natural sources. Marine microorganisms, particularly bacteria of the family
Erythrobacteraceae, have emerged as promising candidates. These bacteria produce carotenoids with demonstrated antiviral and antibacterial properties. Compounds from
Qipengyuania pacifica and related genera have been shown to inhibit viral replication and modulate host inflammatory responses [
10,
11,
12]. Notably, these natural products often exert antiviral effects through mechanisms such as interference with viral genome transcription or modulation of host immune pathways.
Marine bacteria are also advantageous due to their rapid growth in low-cost conditions and ease of large-scale fermentation, making them ideal candidates for sustainable antiviral drug development [
13,
14]. Moreover, carotenoids and other bioactive metabolites derived from marine pigmented bacteria have shown antiviral activity against a wide spectrum of viruses, including herpes simplex virus, IAV, and flaviviruses such as ZIKV and DENV [
15,
16,
17]. Despite these promising findings, the antiviral potential of marine bacteria remains underexplored, particularly in the veterinary virology context.
In this context, our study evaluated the antiviral activity of marine microbial extracts against several enveloped RNA viruses of veterinary importance, including IAV (H1N1, H3N2), influenza B virus (IBV), ZIKV, and DENV2. Among the screened candidates, an extract derived from Parerythrobacter sp. M20A3S10 exhibited significant in vitro antiviral effects with favorable selectivity indices. This strain, belonging to the Erythrobacteraceae family, is expected to produce carotenoid compounds responsible for its antiviral activity. These findings suggest a novel therapeutic potential of marine-derived metabolites, not only as direct-acting antivirals but also as modulators of host immunity and viral pathogenesis.
To date, no studies have reported antiviral activity in Parerythrobacter spp., underscoring the novelty of this study in proposing its application in antiviral research. This work contributes to the growing field at the intersection of viral pathogenesis, immunology, and veterinary epidemiology, offering new insights into host–virus interactions and highlighting marine microbes as promising candidates for next-generation antiviral development.
2. Materials and Methods
2.1. Bacterial Isolation and Culture Conditions
Several bacterial strains, including
Parasphingopyxis,
Roseibium,
Parerythrobacter,
Neorhizobium, and
Qipengyuania species, were collected from seawater in Seosan (Chungnam Province, Republic of Korea) on 16 March 2020. The bacteria from the water sample were cultured as previously described [
17,
18]. The purified strains were routinely cultured on marine agar 2216 (Difco, Franklin Lakes, NJ, USA) at 25 °C and preserved with 20% (
v/
v) glycerol at −80 °C. The bacterial isolates were deposited under the numbers MI00005948, MI00006287, MI00006290, MI00006301, MI00007026, and MI00007049, respectively, at the Microbial Marine BioBank (MMBB) of the National Marine Biodiversity Institute of Korea (MABIK).
2.2. Phylogeny of 16S rRNA Gene Sequences
Genomic DNA was extracted using the Exgene DNA extraction kit (GeneAll, Seoul, Republic of Korea), following the manufacturer’s protocol. The 16S rRNA gene was amplified using bacteria-specific universal primers 27F and 1492R, and sequenced using an automated sequencer (ABI 3730XL, Applied Biosystems, Foster City, CA, USA) at Macrogen Co., Ltd. (Seoul, Republic of Korea) as previously described [
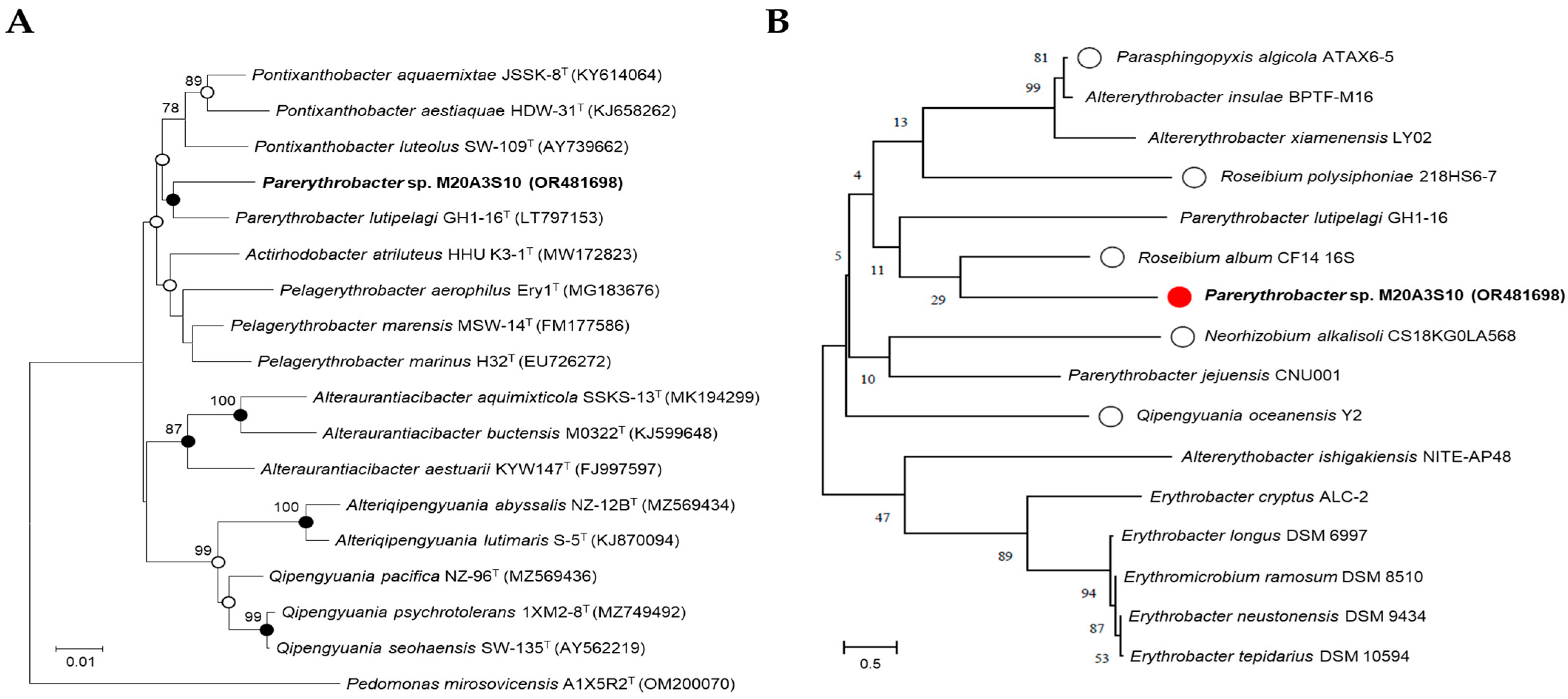
17]. The obtained sequences were assembled with Geneious v9.0.5 to yield a nearly full-length 16S rRNA gene sequence. For taxonomic identification, the 16S rRNA gene sequence (1494 bp) of
Parerythrobacter sp. M20A3S10 was analyzed using the EzBioCloud server (
https://www.ezbiocloud.net/identify, accessed on 30 March 2022) and compared with validated type strains (
Figure 1A). The sequence has been deposited in GenBank under the accession number OR481698.
In addition to this in silico comparison, phylogenetic analysis was also performed among M20A3S10 and other bacterial isolates obtained from the same seawater sample (
Figure 1B), which served as local reference strains. Phylogenetic trees were constructed based on 1419 unambiguously aligned sites using the neighbor-joining (NJ), maximum-likelihood (ML), and maximum-parsimony (MP) methods in MEGA X version 11.0 [
19,
20]. Tree robustness was assessed with 1000 bootstrap replicates across all three algorithms.
2.3. Preparation of the Bacterial Extracts
Bacterial extracts were prepared using a modified method as previously described [
17,
18]. Five bacterial strains—
Parasphingopyxis,
Roseibium,
Parerythrobacter,
Neorhizobium, and
Qipengyuania spp.—were initially cultured in 2.5 L Erlenmeyer flasks containing 1 L of marine broth (total volume: 5 L) under continuous illumination (60 µmol m
−2 s
−1 LED light) at 25 °C. The resulting seed cultures were subsequently transferred to a 20 L panel- or column-type photobioreactor and incubated at a starting concentration of 10
4 CFU mL
−1 under the same conditions for 10–20 days with shaking at 150 rpm. Following cultivation, the culture broth was extracted twice with an equal volume of ethyl acetate (EtOAc). The EtOAc-soluble fractions were pooled and concentrated using a vacuum evaporator. Each bacterial strain yielded approximately 150 mg of crude extract, which was dissolved in dimethyl sulfoxide (DMSO) for subsequent antiviral assays.
2.4. Cells and Viruses
Madin–Darby canine kidney epithelial cells (MDCK; ATCC CCL-34), African green monkey kidney epithelial cells (Vero E6; ATCC CRL-1586), and human lung carcinoma epithelial cells (A549; ATCC CCL-185) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin.
Two strains of IAV and one strain of IBV from ATCC were used: influenza virus A/Puerto Rico/8/34 (H1N1) (A/PR8), A/Wisconsin/15/2009 (H3N2) (A/Wisconsin) strain, and B/Florida/78/2015 Victoria lineage (B/Florida). Both types of flavivirus were provided from the National Culture Collection for Pathogens (Cheongju, Republic of Korea): Dengue virus type 2 (DENV2) isolated from serum samples from Korean patients traveling to Singapore, India, and Thailand in 2015 and Zika virus (ZIKV) strains of Asian/American lineage, PRVABC59.
MDCK cells are widely used for influenza virus propagation due to their high permissiveness, while Vero E6 cells are suitable for the replication of flaviviruses such as ZIKV and DENV. So, the IAV and IBV strains were propagated in MDCK cells supplemented with 1 μg/mL N-tosyl-L-phenylalanine chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich, St. Louis, MO, USA), while ZIKV and DENV were propagated in Vero E6 cells. Viral titers were quantified using a cell culture-based immunofluorescence assay.
2.5. MTT Assay
A confluent monolayer of MDCK and Vero E6 cells was prepared in a 96-well plate at a healthy density of 5.0 × 10
4 cells. The extracts were prepared using a continuous dilution method with an adjusted dilution factor, resulting in the following concentrations: 1000, 500, 250, 100, 50, 25, 10, 5, 2, 1, 0.5, 0.1, 0.01 µg/mL. Cell viability was calculated as a percentage in comparison with DMSO-treated cells using GraphPad Prism software v9.5.1 (GraphPad Software, San Diego, CA, USA) as described previously [
21].
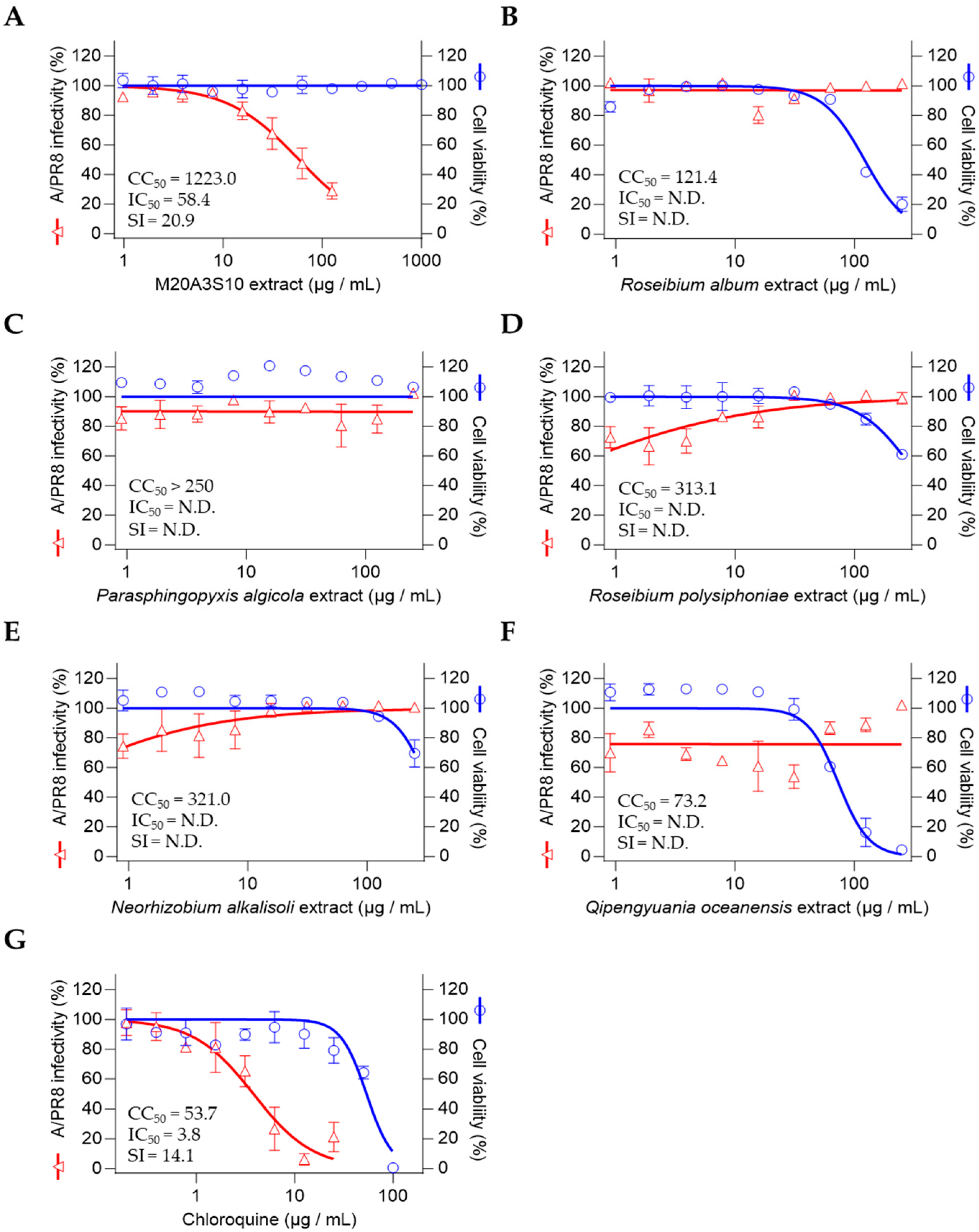
2.6. Preliminary Antiviral Screening
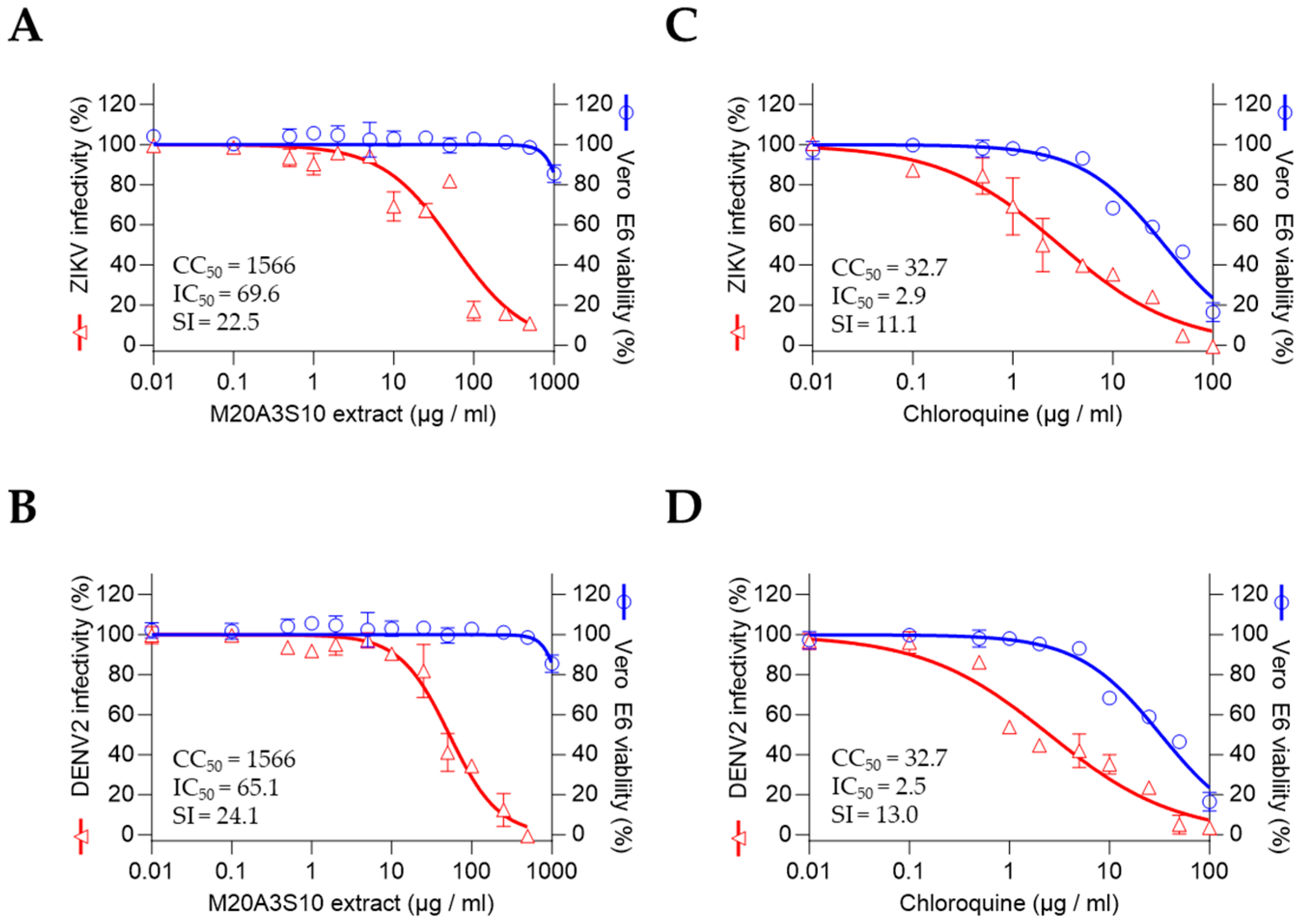
To evaluate antiviral activity, a screening assay was performed using a modified method as previously described [
22,
23]. Permissive cell lines were pretreated with serially diluted bacterial extracts (100, 50, 25, 10, 5, 2, 1, 0.5, 0.1, and 0.01 µg/mL) for 1 h, followed by infection with A/PR8 (H1N1), ZIKV, or DENV2 at a multiplicity of infection (MOI) of 0.1. Viral adsorption was carried out at room temperature for 1 h in a medium supplemented with 1% penicillin–streptomycin and 1 µg/mL TPCK-treated trypsin. After removing the inoculum and washing with Dulbecco’s phosphate-buffered saline (DPBS), the extracts were reapplied and cells were incubated for an additional 48 h. Cell viability was assessed using the MTT assay according to the manufacturer’s instructions [
24], and nonlinear regression analysis was conducted to determine the median inhibitory concentration (IC
50).
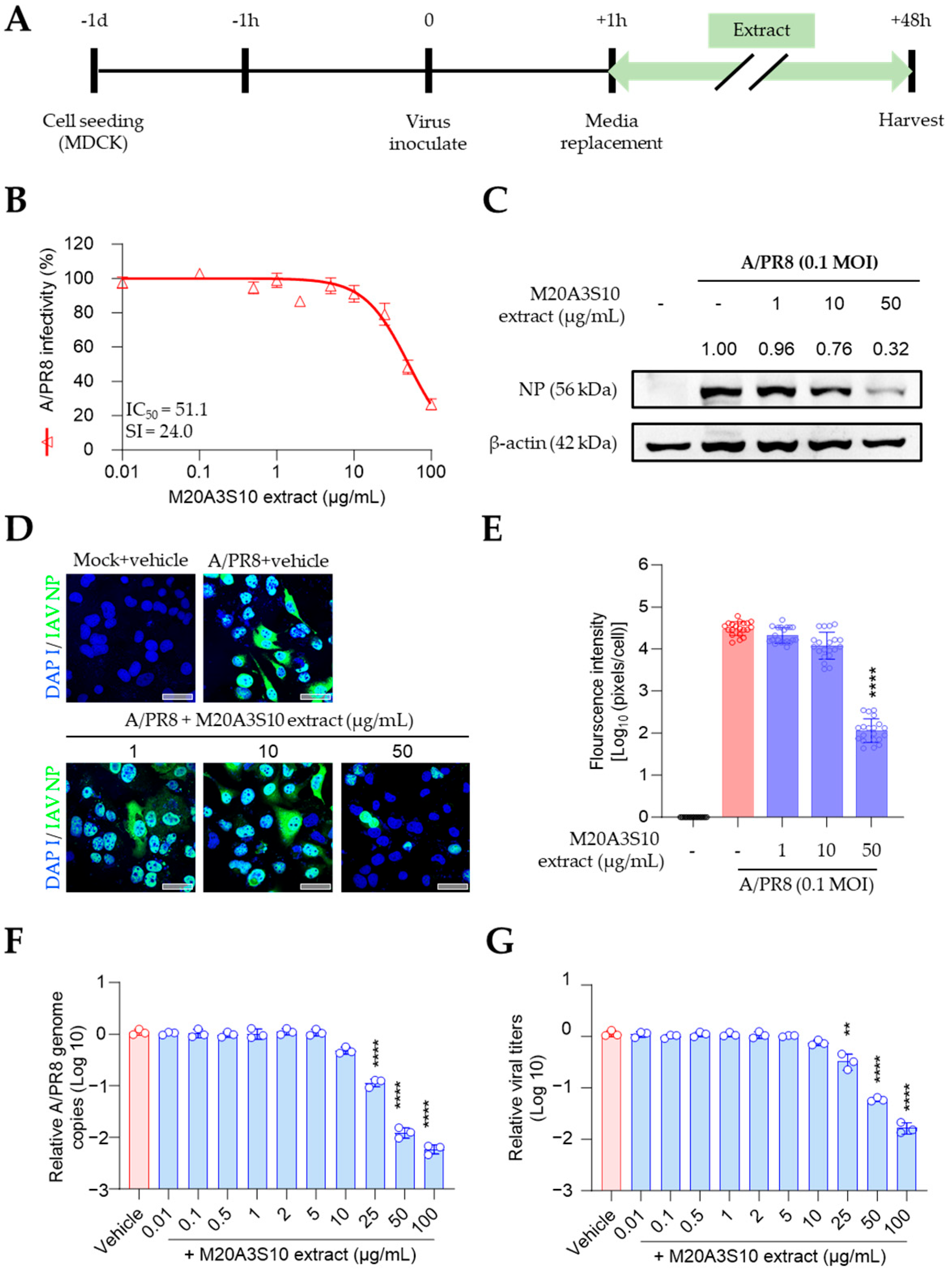
2.7. Time-of-Addition Antiviral Experiment
Time-of-addition antiviral assays were performed to evaluate the stage at which the
Parerythrobacter sp. M20A3S10 extract exerts its antiviral effect. Treatments were administered at three different time points: prior to infection (pre-treatment), concurrently with infection (co-treatment), or after infection (post-treatment), following previously described protocols [
17,
18].
For pre-treatment, cells were incubated with the extract for 1 h, washed with DMEM, and subsequently infected with IAV, ZIKV, or DENV2 at an MOI of 0.1. After 1 h of viral adsorption, cells were washed and replaced with DMEM containing 1 µg/mL TPCK-treated trypsin for IAV, or DMEM alone for ZIKV and DENV2.
For co-treatment, cells were exposed to the virus and extract simultaneously for 1 h at 37 °C, followed by washing and medium replacement as described above.
For post-treatment, cells were first infected with virus for 1 h, washed, and then treated with various concentrations of the extract in DMEM with or without TPCK-treated trypsin, depending on the virus.
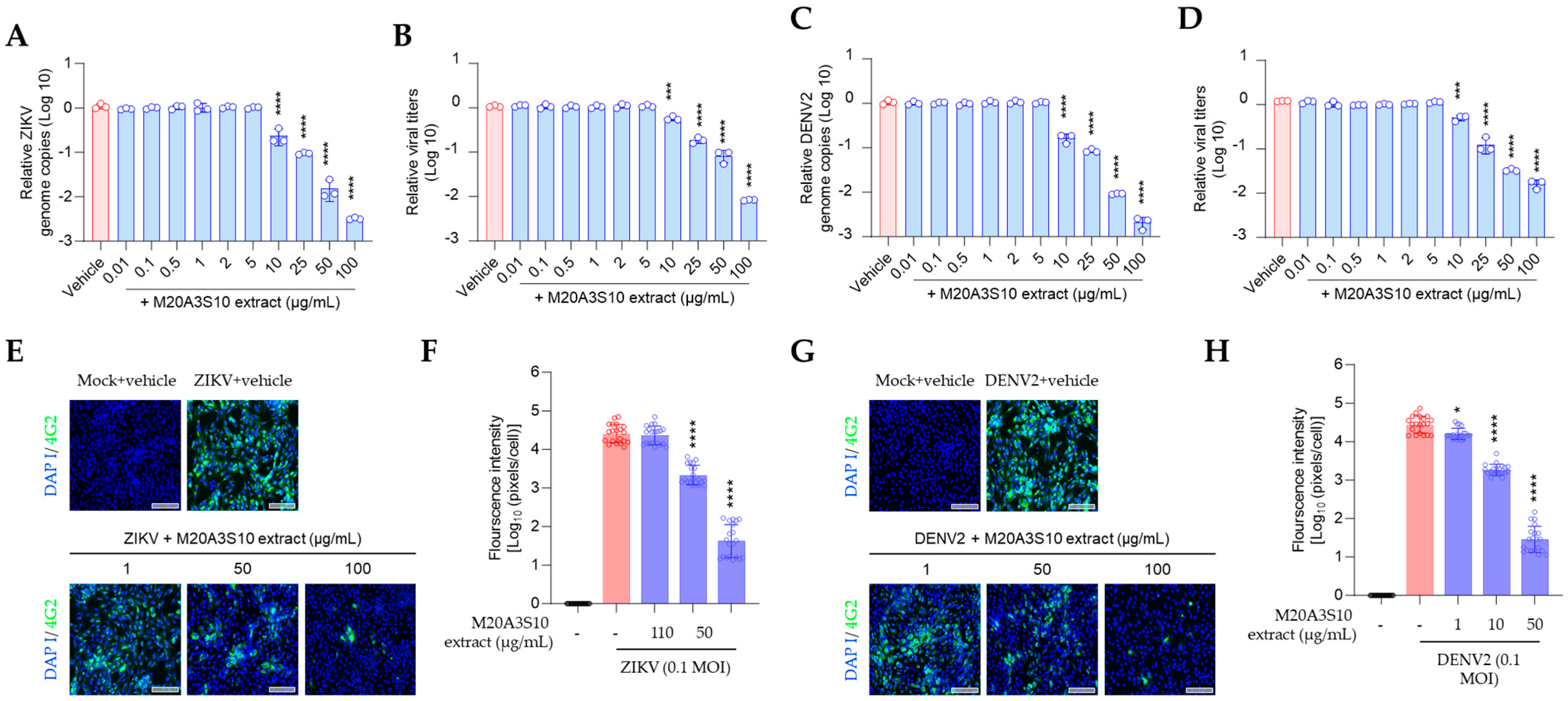
All treatments were performed in triplicate using 96-well plates. At 48 h post-infection, culture supernatants were harvested for the quantification of viral RNA levels and progeny virus titers.
2.8. RT-qPCR
The quantitative RT-qPCR method was used to measure the virus in infected cells, and cell supernatants were analyzed at 48 h after treatment. Virus RNA was extracted using the QIAamp Viral RNA Mini kit (QIAGEN, Hilden, DE, USA) per the manufacturer’s guidelines [
8]. PCR was performed with SensiFAST™ SYBR
® Lo-ROX One-Step Kit (Bioline Meridian BioScience, Cincinnati, OH, USA) as previously described [
17,
18], using the detecting primers as follows: forward (5′-GGCCCTTCAGT TGTTCATC-3′) and reverse primers (5′-GCAGACTTCAGGAATGTG-3′) against IAV PB1, forward (5′-GGTCATGATACTGCTGATTGC-3′) and reverse primers (5′-CCACTAACGTTCTTTTGCA GAC-3′) against ZIKV NS5, forward (5′-AGTTGTT AGTCTRYGTGGACCGAC-3′) and reverse primers (5′-TTGCACCAACAGTCAATGTCTTCAG GTTC-3′) against DENV prM-. The primers were designed in-house using NCBI Primer-BLAST (
https://www.ncbi.nlm.nih.gov/tools/primer-blast, accessed on 15 July 2025) based on conserved regions of the viral genomes.
2.9. Virus Titration
To assess progeny virus production, a 50% tissue culture infective dose (TCID
50) assay was performed. IAV, ZIKV, and DENV titers were determined using their respective permissive cell lines. Briefly, ten-fold serial dilutions of each virus stock were prepared in DMEM, and 100 µL of each dilution was inoculated onto monolayers of permissive cells in 96-well plates. For IAV, the medium contained 1 µg/mL TPCK-treated trypsin; for ZIKV and DENV2, DMEM without trypsin was used. Plates were incubated at 37 °C in a 5% CO
2 atmosphere, and viral cytopathic effects were evaluated at 4 days post-infection. TCID
50/mL values were calculated using the Reed and Muench method [
8].
2.10. Attachment and Penetration Assays
The attachment and penetration assays were performed as described previously [
25]. For the attachment assay, confluent MDCK cells in a 24-well plate were treated with extracts (100 µg/mL) at 4 °C before infection. After 30 min, the inoculum was washed thoroughly with cold DPBS. Then, the A/PR8 strain (1 MOI) was absorbed for 1 h on ice. After thorough washing with cold DPBS, the cells were incubated for 20 h at 37 °C in a 5% CO
2 atmosphere. MDCK cells in a 24-well plate received A/PR8 strain (1 MOI) for 1 h on ice for penetration assay. After thorough washing, the cells were treated with extracts for 10 min and incubated for 20 h at 37 °C in a 5% CO
2 atmosphere.
2.11. Cell Culture Immunofluorescence Assay
MDCK cells were prepared in an 8-well chamber to check viral protein synthesis and infected with the A/PR8 strain (0.1 MOI). Vero E6 cells were also prepared and infected with ZIKV and DENV2 (0.1 MOI) as described elsewhere [
8]. After fixation with 4% PFA for 10 min at RT, the chamber was washed and blocked with 5% Bovine Serum Albumin (BSA) at RT for 1 h to reduce non-specific reaction.
Subsequently, cells were incubated overnight at 4 °C with primary antibodies targeting either the influenza A virus M2 protein (ab5416, Abcam, Cambridge, UK) or the flavivirus group envelope protein (clone 4G2, Native Antigen Company, Oxfordshire, UK). The next day, after washing, a fluorescently labeled secondary antibody (goat anti-mouse IgG conjugated with Alexa Fluor (AF) 488; A-11001, Thermo Scientific, Waltham, MA, USA) was applied and incubated for 1 h at RT. Fluorescence images were captured using a fluorescence microscope to assess the presence and distribution of viral proteins.
2.12. Western Blotting
To evaluate the expression levels of viral proteins in cultured cells, Western blot analysis was performed as previously described [
8]. Briefly, cells were lysed using RIPA buffer containing 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na
2P
2O
7, 2 mM Na
3VO
4, 1% Triton X-100, 10% glycerol, 0.1% SDS, and 0.5% deoxycholate (Invitrogen, Waltham, MA, USA) for 10 min on ice. The lysates were centrifuged at 12,000×
g for 10 min at 4 °C, and the supernatant was collected. Total protein concentration was determined using a BCA protein assay kit (Thermo Scientific).
Equal amounts of protein were subjected to SDS-PAGE and transferred onto nitrocellulose membranes (GE Healthcare Life Sciences, Lafayette, CO, USA). The membranes were blocked for 1 h at room temperature in Tris-buffered saline containing 5% skim milk and 0.1% Tween-20, then incubated overnight at 4 °C with a primary antibody against IAV NP (ab128193, Abcam, Cambridge, UK) or β-actin (sc-47778, Santa Cruz Biotechnology, Dallas, TX, USA). After washing, membranes were incubated for 1 h at room temperature with HRP-conjugated goat anti-mouse IgG secondary antibody (SC-2005, Santa Cruz Biotechnology, Dallas, TX, USA). Protein bands were visualized using enhanced chemiluminescence (ECL) substrate (Dogen, Seoul, Republic of Korea) and imaged with a Davinch-K imaging system (Youngwha Scientific Co., Ltd., Seoul, Republic of Korea). Band intensities were quantified and normalized to β-actin as an internal control, and the results were expressed as relative fold changes.
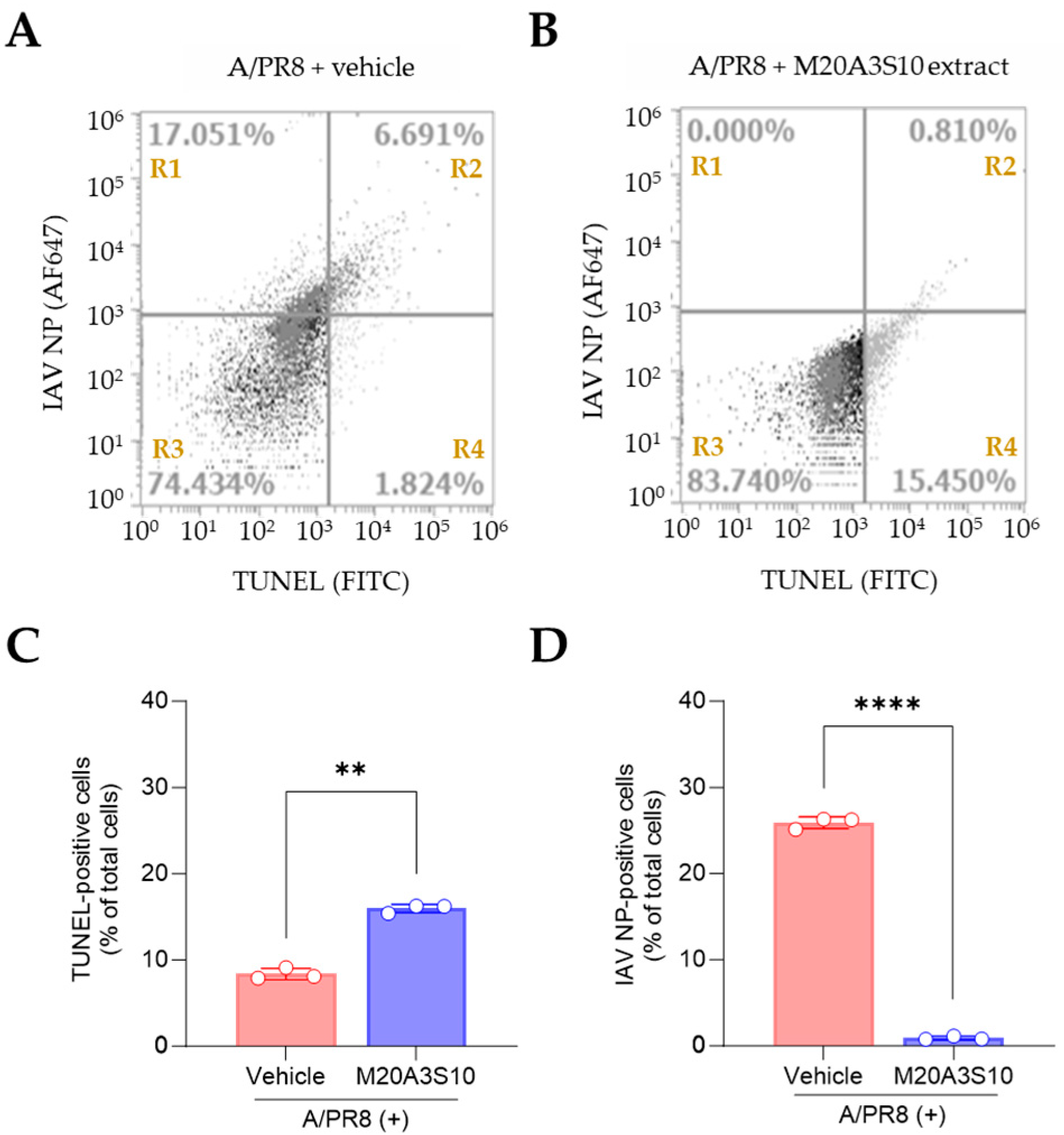
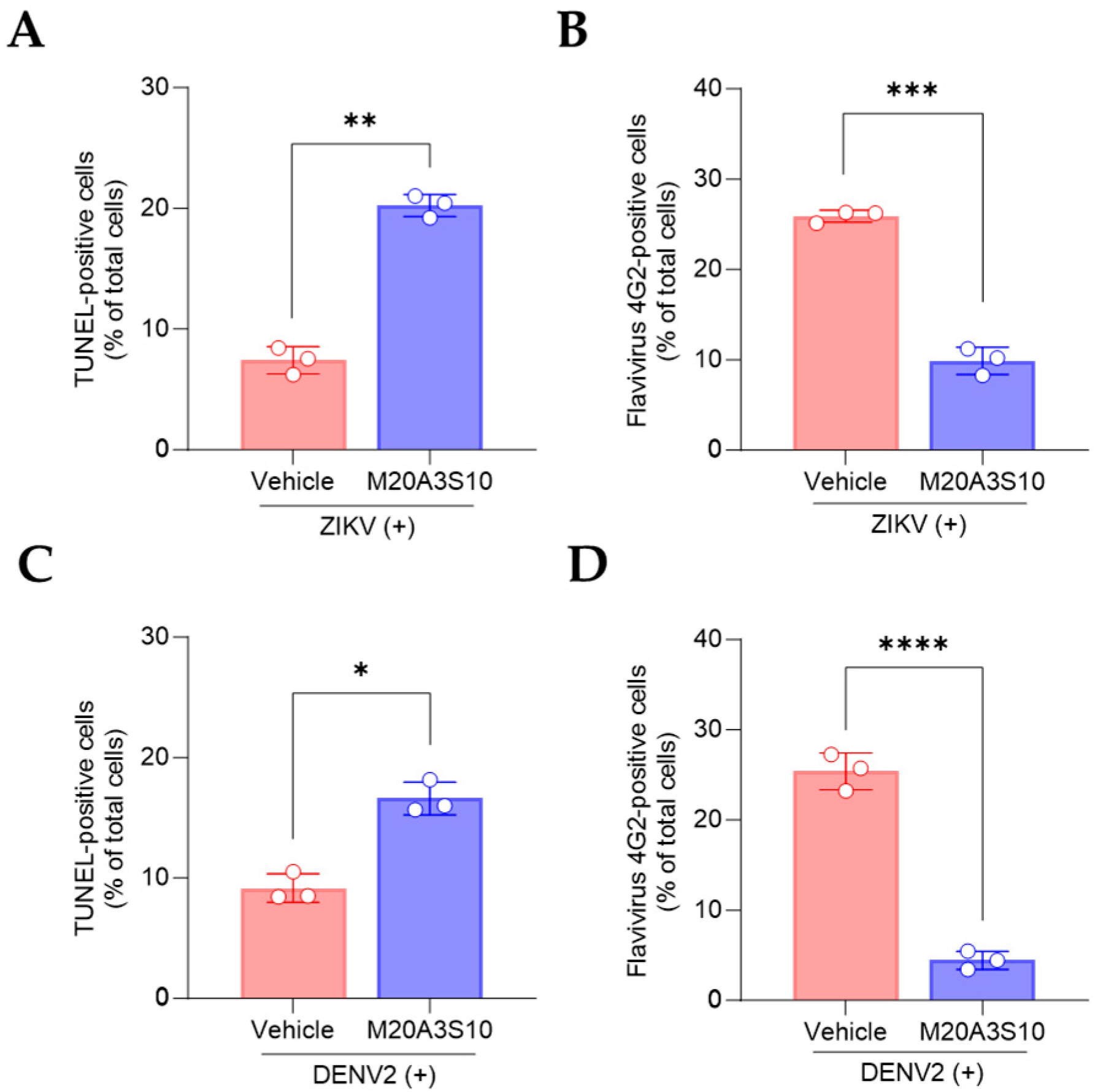
2.13. Flow Cytometry
The present study utilized flow cytometry to assess the induction of viral protein and apoptosis. The methodology adopted was in accordance with previously established protocols [
17]. Briefly, MDCK cells were inoculated with A/PR8 strain (0.1 MOI), and Vero E6 cells were inoculated with ZIKV (0.1 MOI) or DENV2 (0.1 MOI). Primary antibodies against IAV NP (ab128193, Abcam, Cambridge, UK) and flavivirus envelope protein (4G2, Native Antigen Company, Oxford, UK) and secondary antibodies conjugated with goat-anti mouse AF647 (A32728, Thermo Scientific) were applied to evaluate viral protein. For apoptosis detection, a TUNEL assay was performed using the In Situ Cell Death Detection Kit (11684795910, Roche, Basel, Switzerland) according to the manufacturer’s instructions. Flow cytometry was performed using the AttuneTM NxT flow cytometer (Thermo Fisher Scientific), and the AttuneTM NxT software v3.1.2 was used to digitize the data from each sample.
2.14. Statistical Analysis
Statistical analysis was carried out using GraphPad Prism software, with one-way ANOVA being employed. The data was expressed as the mean ± standard deviation (SD) of at least three independent experiments. Statistical significance was considered at the following levels: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. The selective index (SI) was calculated using sigmoidal dose–response curves, with the following equation: SI = mean CC50/mean IC50. The ratio of CC50 to IC50, i.e., CC50/IC50, was employed to calculate SI.
4. Discussion
The ongoing threat of zoonotic RNA viruses such as IAV, ZIKV, and DENV highlights the urgent need for novel antiviral agents with broad-spectrum efficacy. These viruses exhibit high mutation rates, broad host ranges, and complex interactions with host immune systems, contributing to repeated outbreaks and significant veterinary and public health burdens. In this study, we evaluated the antiviral potential of a marine bacterial extract derived from Parerythrobacter sp. M20A3S10, isolated from coastal seawater. The extract exhibited notable in vitro antiviral activity against a diverse group of enveloped RNA viruses of veterinary importance, including IAV (H1N1, H3N2), IBV, ZIKV, and DENV2, suggesting a broad-spectrum mechanism of action.
From a veterinary immunology and pathogenesis perspective, a key finding was the extract’s capacity to inhibit viral replication by promoting apoptosis-mediated antiviral responses. This is particularly significant in the context of host–pathogen interactions: RNA viruses often hijack host cell death pathways to facilitate replication, shifting host cells toward necroptosis, which supports progeny production and inflammation [
7,
8]. Our data support the hypothesis that modulating this balance toward apoptosis—a more controlled form of cell death—can restrict viral replication and enhance phagocytic clearance of infected cells [
6]. This insight provides a valuable immunological target for therapeutic intervention, especially for veterinary viruses that compromise immune organs and epithelial barriers.
Notably, post-treatment with the M20A3S10 extract was effective against all tested viruses, whereas pre- and co-treatment failed to prevent infection. Antiviral mechanisms associated with pre-treatment typically involve direct viral inactivation or interference with viral attachment to host cells. In contrast, co-treatment generally targets early stages of infection such as viral entry, membrane fusion, or endocytosis. However, neither pre- nor co-treatment suppressed viral replication, while post-treatment significantly reduced infection. These findings suggest that the antiviral effects of the extract are predominantly exerted at post-entry stages, likely during viral genome replication, viral protein synthesis, or interactions with host immune surveillance mechanisms.
This temporal pattern aligns with the activation of host apoptotic responses and implies that the extract’s active components may function by sensitizing infected cells to apoptosis or enhancing caspase-dependent cell death pathways [
17]. Such a mechanism is particularly relevant, as apoptosis-mediated immune clearance represents a crucial antiviral defense in both human and veterinary hosts and serves as a promising therapeutic target in veterinary virology [
31].
The extract’s efficacy was consistent across multiple influenza strains and cell types, including human and canine epithelial cells, which are representative of major target tissues in both human and veterinary infections. This cross-strain and cross-species efficacy supports the potential of this marine-derived compound as a versatile antiviral agent. Furthermore, its superior selectivity index compared to chloroquine indicates a more favorable therapeutic window, encouraging further pharmacological development.
To date, no studies have reported the antiviral activity of
Parerythrobacter spp., underscoring the novelty of this study in exploring its potential as an antiviral agent. Although the specific active compound responsible for the observed effect remains unidentified, genomic analysis of
Parerythrobacter aurantius has revealed the presence of a complete carotenoid biosynthesis gene cluster [
32]. This finding supports the likelihood that
Parerythrobacter spp. can produce bioactive pigments—such as carotenoids—with multifunctional properties, including potential antiviral and antioxidant effects.
In the context of epidemiology, the rising incidence of zoonotic and vector-borne viral diseases is exacerbated by climate change, wildlife–livestock–human interface expansion, and globalized movement of animals and goods. Natural compounds from marine microbes represent an underexploited resource for pandemic preparedness. Although
Parerythrobacter is known to produce carotenoids with reported antiviral, anti-inflammatory, and antioxidative properties [
11,
33], the contribution of these compounds to the observed antiviral effects in this study remains hypothetical. Currently, there is no direct evidence linking the carotenoids present in the extract to its antiviral mechanism. Future studies should include pure compound isolation, validation, and gene knockout assays to confirm the active constituents. As the active components remain uncharacterized, it is also possible that other metabolites may contribute to the antiviral activity, either through direct inhibition of viral replication or by modulating the host immune environment. These findings highlight the need for further mechanistic investigations to elucidate both the molecular targets and the bioactive constituents involved.
Although the precise molecular constituents responsible for the observed effects remain to be identified, the data suggest that components of the extract—potentially carotenoids—interfere with late-stage viral replication and promote host-driven clearance mechanisms. Further metabolomic characterization (e.g., via LC-MS/MS) and in vivo efficacy testing in relevant animal models are warranted. Such studies could determine pharmacokinetics, bioavailability, and host-specific responses, which are essential for translational application in veterinary medicine. These experiments will begin using the A/PR8 (H1N1) strain, given its well-established utility in both in vitro and in vivo models [
8]. In the future, additional studies leveraging the broad replication capacity of this virus may provide further insights into the antiviral potential of the extract.
Marine bacteria were chosen for this investigation due to their rich metabolic diversity, rapid growth in low-cost media, and proven capacity to produce antiviral pigments such as carotenoids [
13,
14]. While specific compounds were not isolated in this study, we aimed to showcase the scalability and feasibility of this resource. Notably, this study integrated a single screening platform to evaluate antiviral activity against IAV, IBV, ZIKV, and DENV2—viruses of both veterinary and zoonotic importance. Furthermore, flow cytometry-based dual quantification of viral protein and host apoptosis provided a mechanistic dimension rarely explored in previous studies.
In conclusion, the findings underscore the value of apoptosis modulation as an antiviral strategy and identify marine carotenoid-producing bacteria as promising candidates for the development of next-generation antivirals. Given the cross-species and broad-spectrum efficacy observed, this approach holds potential not only for companion animals and livestock but also for wildlife disease management and zoonotic outbreak containment.
5. Conclusions
In this study, we identified a marine bacterial extract from Parerythrobacter sp. M20A3S10 that exhibits broad-spectrum in vitro antiviral activity against several enveloped RNA viruses of veterinary and zoonotic concern, including IAV, IBV, ZIKV, and DENV. The extract was most effective when administered post-infection, suggesting that its antiviral action may involve interference with viral replication or modulation of host cell responses. The ability to inhibit viral proliferation in both human and canine epithelial cells underscores its potential relevance for veterinary antiviral development.
Although the precise bioactive constituents remain unidentified, bacteria of the Erythrobacteraceae family are known to produce compounds such as carotenoids with reported antiviral properties. While the presence of such metabolites in the tested extract has not yet been confirmed, their potential contribution warrants further investigation. These findings support the continued exploration of marine microbial resources as candidates for novel antiviral strategies, and future studies should focus on compound isolation, mechanistic analysis, and in vivo validation in relevant animal models.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}