
Genetic Diversity and Phylogenetic Relationships of Castor fiber birulai in Xinjiang, China, Revealed by Mitochondrial Cytb and D-loop Sequence Analyses

 and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Amplification of Cytb and D-Loop Genes
2.3. DNA Sequence Data Processing
3. Results
3.1. Mitochondrial Sequence Analysis
3.2. Genetic Diversity of C. f. birulai
3.3. Genetic Structure of Xinjiang C. f. birulai
3.4. Haplotype Network Analysis
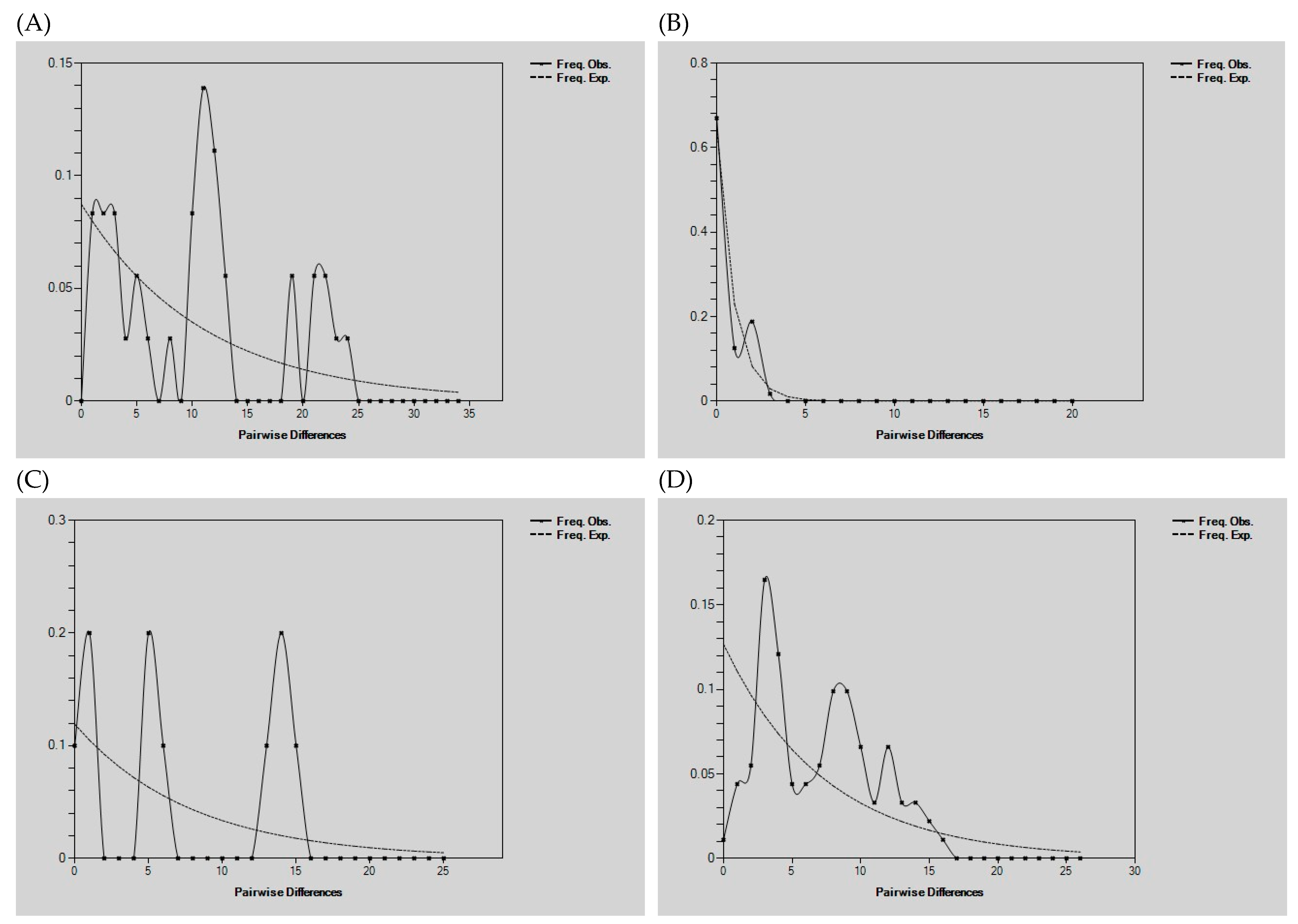
3.5. Demographic History Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosell, F.; BozsÉR, O.; Collen, P.; Parker, H. Ecological impact of beavers Castor fiber and Castor canadensis and their ability to modify ecosystems. Mammal Rev. 2005, 35, 248–276. [Google Scholar] [CrossRef]
- Biedrzycka, A.; Konior, M.; Babik, W.; Świsłocka, M.; Ratkiewicz, M. Admixture of two phylogeographic lineages of the Eurasian beaver in Poland. Mamm. Biol. 2014, 79, 287–296. [Google Scholar] [CrossRef]
- Halley, D.J.; Saveljev, A.P.; Rosell, F. Population and distribution of beavers Castor fiber and Castor canadensis in Eurasia. Mammal Rev. 2021, 51, 1–24. [Google Scholar] [CrossRef]
- Falaschi, M.; Ficetola, G.F.; Viviano, A.; Mazza, G.; Mori, E. Environmental suitability and potential range expansion of the Eurasian beaver in Italy. Anim. Conserv. 2023, 27, 324–337. [Google Scholar] [CrossRef]
- Gaywood, M.J. Reintroducing the Eurasian beaver Castor fiber to Scotland. Mammal Rev. 2017, 48, 48–61. [Google Scholar] [CrossRef]
- Halley, D.J. Sourcing Eurasian beaver Castor fiber stock for reintroductions in Great Britain and Western Europe. Mammal Rev. 2010, 41, 40–53. [Google Scholar] [CrossRef]
- Marr, M.M.; Brace, S.; Schreve, D.C.; Barnes, I. Identifying source populations for the reintroduction of the Eurasian beaver, Castor fiber L. 1758, into Britain: Evidence from ancient DNA. Sci. Rep. 2018, 8, 2708. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Palmer, R.; Rosell, F.; Naylor, A.; Cole, G.; Mota, S.; Brown, D.; Fraser, M.; Pizzi, R.; Elliott, M.; Wilson, K.; et al. Eurasian beaver (Castor fiber) health surveillance in Britain: Assessing a disjunctive reintroduced population. Vet. Rec. 2021, 188, e84. [Google Scholar] [CrossRef] [PubMed]
- Senn, H.; Ogden, R.; Frosch, C.; Syrůčková, A.; Campbell-Palmer, R.; Munclinger, P.; Durka, W.; Kraus, R.H.S.; Saveljev, A.P.; Nowak, C.; et al. Nuclear and mitochondrial genetic structure in the Eurasian beaver (Castor fiber)—Implications for future reintroductions. Evol. Appl. 2014, 7, 645–662. [Google Scholar] [CrossRef] [PubMed]
- Ducroz, J.-F.; Stubbe, M.; Saveljev, A.P.; Heidecke, D.; Samjaa, R.; Ulevičius, A.; Stubbe, A.; Durka, W. Genetic Variation and Population Structure of the Eurasian Beaver Castor fiber in Eastern Europe and Asia. J. Mammal. 2005, 86, 1059–1067. [Google Scholar] [CrossRef]
- Chu, H.; Jiang, Z. Distribution and conservation of the Sino-Mongolian beaver Castor fiber birulai in China. Oryx 2009, 43, 197–202. [Google Scholar] [CrossRef]
- Saveljev, A.P.; Niamosor, B.; Shaariybuu, B.; Batseren, D. Self-eating in beavers—Trophic opportunism or reaction on stress Extreme case from Mongolia. Russ. J. Theriol. 2016, 15, 68–74. [Google Scholar] [CrossRef]
- Halley, D.; Rosell, F.; Saveljev, A. Population and distribution of Eurasian beaver (Castor fiber). Balt. For. 2012, 18, 168–175. [Google Scholar]
- Horn, S.; Durka, W.; Wolf, R.; Ermala, A.; Stubbe, A.; Stubbe, M.; Hofreiter, M. Mitochondrial genomes reveal slow rates of molecular evolution and the timing of speciation in beavers (Castor), one of the largest rodent species. PLoS ONE 2011, 6, e14622. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer Premier: Program for Design of Degenerate Primers from a Protein Sequence. BioTechniques 2018, 24, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, X.; Gai, J.; Yu, D. Isolation and characterization of a seed-specific isoform of microsomal omega-6 fatty acid desaturase gene (FAD2-1B) from soybean: Full Length Research Article. DNA Seq. 2008, 19, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Burland, T.G. DNASTAR’s Lasergene Sequence Analysis Software; Humana Press: Totowa, NJ, USA, 1999; pp. 71–91. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S.; Battistuzzi, F.U. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimatingf-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Exeoeffr, L.; Smouse, P.; Quattro, J.J.G. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Applications to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Moritz, C. Defining ‘evolutionarily significant units’ for conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Hoban, S.; Bruford, M.W.; da Silva, J.M.; Funk, W.C.; Frankham, R.; Gill, M.J.; Grueber, C.E.; Heuertz, M.; Hunter, M.E.; Kershaw, F.; et al. Genetic diversity goals and targets have improved, but remain insufficient for clear implementation of the post-2020 global biodiversity framework. Conserv. Genet. 2023, 24, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Sheikh, S.I.; Ward, A.K.G.; Forbes, A.A.; Prior, K.M.; Stone, G.N.; Gates, M.W.; Egan, S.P.; Zhang, L.; Davis, C.; et al. Delimiting the cryptic diversity and host preferences of Sycophila parasitoid wasps associated with oak galls using phylogenomic data. Mol. Ecol. 2022, 31, 4417–4433. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Loire, E.; Chiari, Y.; Bernard, A.; Cahais, V.; Romiguier, J.; Nabholz, B.; Lourenço, J.M.; Galtier, N.J.G.B. Population genomics of the endangered giant Galápagos tortoise. Genome Biol. 2013, 14, R136. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chang, J.; Han, L.; Liu, T.; Li, G.; Garber, P.A.; Xiao, N.; Zhou, J. The Genetic Status of the Critically Endangered Hainan Gibbon (Nomascus hainanus): A Species Moving Toward Extinction. Front. Genet. 2020, 11, 608633. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.; Zinner, D.; Li, Y.; Yao, X.; Roos, C.; Yu, L. Recent Advances in Genetics and Genomics of Snub-Nosed Monkeys (Rhinopithecus) and Their Implications for Phylogeny, Conservation, and Adaptation. Genes 2023, 14, 985. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Morán, J.; Saavedra, D.; Manteca-Vilanova, X. Reintroduction of the Eurasian otter (Lutra lutra) in northeastern Spain: Trapping, handling, and medical management. J. Zoo Wildl. Med. 2002, 33, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Canestrelli, D.; Frosch, C.; Kraus, R.H.S.; Angst, C.; Allgöwer, R.; Michaux, J.; Teubner, J.; Nowak, C. The Genetic Legacy of Multiple Beaver Reintroductions in Central Europe. PLoS ONE 2014, 9, e97619. [Google Scholar] [CrossRef]
- Tigano, A.; Friesen, V.L. Genomics of local adaptation with gene flow. Mol. Ecol. 2016, 25, 2144–2164. [Google Scholar] [CrossRef] [PubMed]
- Gouskov, A.; Vorburger, C. Postglacial recolonizations, watershed crossings and human translocations shape the distribution of chub lineages around the Swiss Alps. BMC Evol. Biol. 2016, 16, 185. [Google Scholar] [CrossRef] [PubMed]
- Caramelli, D.; Elsner, J.; Hofreiter, M.; Schibler, J.; Schlumbaum, A. Ancient mtDNA diversity reveals specific population development of wild horses in Switzerland after the Last Glacial Maximum. PLoS ONE 2017, 12, e0177458. [Google Scholar] [CrossRef]
- Groombridge, J.J.; Raisin, C.; Bristol, R.; Richardson, D.S. Genetic consequences of reintroductions and insights from population history. In Reintroduction Biology: Integrating Science and Management; Wiley: Oxford, UK, 2012; pp. 395–440. [Google Scholar]
- Preatoni, D.; Mustoni, A.; Martinoli, A.; Carlini, E.; Chiarenzi, B.; Chiozzini, S.; Van Dongen, S.; Wauters, L.A.; Tosi, G. Conservation of brown bear in the Alps: Space use and settlement behavior of reintroduced bears. Acta Oecologica 2005, 28, 189–197. [Google Scholar] [CrossRef]
- Swenson, J.E.; Taberlet, P.; Bellemain, E. Genetics and conservation of European brown bears Ursus arctos. Mammal Rev. 2011, 41, 87–98. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, T.; Li, Y.; Xu, S.; Zhang, M.; Hu, X.; Liu, S.; Hu, D.; Wronski, T. Identifying personality traits and their potential application to the management of captive forest musk deer (Moschus berezovskii). Appl. Anim. Behav. Sci. 2021, 234, 105168. [Google Scholar] [CrossRef]
- Tarav, M.; Tokunaga, M.; Kondo, T.; Kato-Mori, Y.; Hoshino, B.; Dorj, U.; Hagiwara, K. Problems in the Protection of Reintroduced Przewalski’s Horses (Equus ferus przewalskii) Caused by Piroplasmosis. J. Wildl. Dis. 2017, 53, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Geue, J.C.; Bertola, L.D.; Bloomer, P.; Brüniche-Olsen, A.; da Silva, J.M.; DeWoody, J.A.; Fedorca, A.; Godoy, J.A.; Grueber, C.E.; Hunter, M.E.; et al. Practical genetic diversity protection: An accessible framework for IUCN subpopulation and Evolutionarily Significant Unit identification. EcoEvoRxiv, 2025; preprint. [Google Scholar] [CrossRef]
- Crandall, K.A.; Bininda-Emonds, O.R.P.; Mace, G.M.; Wayne, R.K. Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 2000, 15, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.E.; Klop-Toker, K.; Wallace, S.; Kelly, O.; Callen, A.; Seeto, R.; Mahony, S.V.; Hayward, M.W.; Mahony, M.J. Uncovering inbreeding, small populations, and strong genetic isolation in an Australian threatened frog, Litoria littlejohni. Conserv. Genet. 2023, 24, 575–588. [Google Scholar] [CrossRef]
- Perrin, A.; Khimoun, A.; Faivre, B.; Ollivier, A.; de Pracontal, N.; Théron, F.; Loubon, M.; Leblond, G.; Duron, O.; Garnier, S. Habitat fragmentation differentially shapes neutral and immune gene variation in a tropical bird species. Heredity 2021, 126, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Yang, S.; Li, H.; Zhang, Y.; Li, R.; Sahu, S.K.; Deng, W.; Liu, B.; Shi, M.; Wang, S.; et al. Large-scale genome sequencing of giant pandas improves the understanding of population structure and future conservation initiatives. Proc. Natl. Acad. Sci. USA 2024, 121, e2406343121. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Burley, J.T.; Liu, Y.; Chang, J.; Chen, D.; Lu, Q.; Li, S.-H.; Zhou, X.; Edwards, S.; Zhang, Z.; et al. Genomic Consequences of Long-Term Population Decline in Brown Eared Pheasant. Mol. Biol. Evol. 2021, 38, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.A.; Moilanen, A.; Wintle, B.A.; Thomas, C.D. Habitat area, quality and connectivity: Striking the balance for efficient conservation. J. Appl. Ecol. 2010, 48, 148–152. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Bp | Haplotypes | Sample |
|---|---|---|---|
| Cytb | 800 | \ | \ |
| D-loop | 592 | Hap_1 | L_1, L_2_1, L_2_2, L_3_1, L_3_2, L_3_3, L_3_4, L_3_5, L_4, L_5_2, M_2, M_3, M_4, M_5, M_6 |
| Hap_2 | L_5_1, L_5_3 | ||
| Hap_3 | L_6, M_1, M_2 | ||
| Cytb + D-loop | 1258 | Hap_1 | L_1, L_2_1, L_2_2, L_3_1, L_3_2, L_3_3, L_3_4, L_3_5, L_4, L_5_2, M_2, M_3, M_4, M_5, M_6 |
| Hap_2 | L_5_1, L_5_3 | ||
| Hap_3 | L_6, M_1, M_2 |
| Population | H | π | K | |||
|---|---|---|---|---|---|---|
| Cytb | D-loop | Cytb | D-loop | Cytb | D-loop | |
| China-C. f. birulai | 1 | 0.576 | \ | 0.00515 | \ | 2.41558 |
| Mongolia-C. f. birulai | 2 | 0.600 | 0.00083 | 0.00248 | 0.667 | 1.20 |
| Castor fiber | 7 | 0.967 | 0.00411 | 0.02123 | 3.289 | 8.15398 |
| Castor canadensis | 3 | 0.989 | 0.00440 | 0.01472 | 3.500 | 6.89011 |
| Country | Subspecies | C. f. pohlei | mogolin_ C. f. briulai | C. f. tuvinivus | C. f. albicus | C. f. fiber | C. f. galliae | China_ C. f. briulai | C. f. belorussicus | Castor canadensis |
|---|---|---|---|---|---|---|---|---|---|---|
| Russia | C. f. pohlei | - | 0.86900 | 0.95059 | 0.94203 | 0.89286 | 0.89831 | 0.97056 | 0.90323 | 0.93333 |
| Mogolin | mogolin_ C. f. briulai | 0.02389 | - | 0.83434 | 0.95033 | 0.94059 | 0.93750 | 0.67055 | 0.93407 | 0.93627 |
| Russia | C. f. tuvinivus | 0.02357 | 0.02826 | - | 0.90330 | 0.84158 | 0.85965 | 0.95059 | 0.85321 | 0.93368 |
| Germany | C. f. albicus | 0.05007 | 0.04446 | 0.05262 | - | 0.95349 | 0.87500 | 0.98880 | 0.84615 | 0.93267 |
| Norway | C. f. fiber | 0.04022 | 0.04370 | 0.04367 | 0.03063 | - | NA | 0.99093 | NA | 0.92314 |
| French | C. f. galliae | 0.04248 | 0.04147 | 0.04958 | 0.01118 | 0.02770 | - | 0.99043 | NA | 0.92350 |
| China | China_ C. f. briulai | 0.02494 | 0.00269 | 0.03002 | 0.04200 | 0.04125 | 0.03902 | - | 0.98987 | 0.96743 |
| Russia | C. f. belorussicus | 0.04474 | 0.03919 | 0.04727 | 0.00906 | 0.02990 | 0.01047 | 0.03676 | - | 0.91914 |
| Canada | Castor canadensis | 0.24048 | 0.21939 | 0.23211 | 0.22851 | 0.23189 | 0.23341 | 0.21630 | 0.21754 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Ma, Y.; He, C.; Huang, C.; Gao, X.; Ding, P.; Zhong, L. Genetic Diversity and Phylogenetic Relationships of Castor fiber birulai in Xinjiang, China, Revealed by Mitochondrial Cytb and D-loop Sequence Analyses. Animals 2025, 15, 2096. https://doi.org/10.3390/ani15142096
Zhu L, Ma Y, He C, Huang C, Gao X, Ding P, Zhong L. Genetic Diversity and Phylogenetic Relationships of Castor fiber birulai in Xinjiang, China, Revealed by Mitochondrial Cytb and D-loop Sequence Analyses. Animals. 2025; 15(14):2096. https://doi.org/10.3390/ani15142096
Chicago/Turabian StyleZhu, Linyin, Yingjie Ma, Chengbin He, Chuang Huang, Xiaobo Gao, Peng Ding, and Linqiang Zhong. 2025. "Genetic Diversity and Phylogenetic Relationships of Castor fiber birulai in Xinjiang, China, Revealed by Mitochondrial Cytb and D-loop Sequence Analyses" Animals 15, no. 14: 2096. https://doi.org/10.3390/ani15142096
APA StyleZhu, L., Ma, Y., He, C., Huang, C., Gao, X., Ding, P., & Zhong, L. (2025). Genetic Diversity and Phylogenetic Relationships of Castor fiber birulai in Xinjiang, China, Revealed by Mitochondrial Cytb and D-loop Sequence Analyses. Animals, 15(14), 2096. https://doi.org/10.3390/ani15142096