
In Silico Identification of Novel Compounds as Anthelmintics Against Haemonchus contortus Through Inhibiting β-Tubulin Isotype 1 and Glutathione S-Transferase


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Structural Prediction and Protein Preparation
2.2. Pharmacophore Modeling and Virtual Screening
2.3. Lead Compound Docking
2.4. ADMET Analysis
2.5. Molecular Dynamics (MD) Simulations
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Charlier, J.; Rinaldi, L.; Musella, V.; Ploeger, H.W.; Chartier, C.; Vineer, H.R.; Hinney, B.; von Samson-Himmelstjerna, G.; Băcescu, B.; Mickiewicz, M. Initial assessment of the economic burden of major parasitic helminth infections to the ruminant livestock industry in Europe. Prev. Vet. Med. 2020, 182, 105103. [Google Scholar] [CrossRef] [PubMed]
- Besier, R.; Kahn, L.; Sargison, N.; Van Wyk, J.A. The pathophysiology, ecology and epidemiology of Haemonchus contortus infection in small ruminants. Adv. Parasitol. 2016, 93, 95–143. [Google Scholar] [PubMed]
- Kaplan, R.M.; Vidyashankar, A.N. An inconvenient truth: Global worming and anthelmintic resistance. Vet. Parasitol. 2012, 186, 70–78. [Google Scholar] [CrossRef]
- Britton, C.; Roberts, B.; Marks, N. Functional genomics tools for Haemonchus contortus and lessons from other helminths. Adv. Parasitol. 2016, 93, 599–623. [Google Scholar]
- Cvilink, V.; Lamka, J.; Skálová, L. Xenobiotic metabolizing enzymes and metabolism of anthelminthics in helminths. Drug Metab. Rev. 2009, 41, 8–26. [Google Scholar] [CrossRef]
- Munguía, B.; Teixeira, R.; Veroli, V.; Melian, E.; Saldaña, J.; Minteguiaga, M.; Señorale, M.; Marín, M.; Domínguez, L. Purification of native M. vogae and H. contortus tubulin by TOG affinity chromatography. Exp. Parasitol. 2017, 182, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sainas, S.; Dosio, F.; Boschi, D.; Lolli, M.L. Targeting human onchocerciasis: Recent advances beyond ivermectin. Annu. Rep. Med. Chem. 2018, 51, 1–38. [Google Scholar]
- Shalaby, H.A. Anthelmintics resistance; how to overcome it? Iran. J. Parasitol. 2013, 8, 18. [Google Scholar]
- Sivachandran, R.; Lakshmi, K.N.; Sivamurugan, V.; Priya, P. Effect of Punica granatum on the Glutathione S-Transferase Activity of Haemonchus Contortus-An In Vitro And In Silico Analyses. Uttar Pradesh J. Zool. 2021, 42, 475–485. [Google Scholar]
- Harder, A. The biochemistry of Haemonchus contortus and other parasitic nematodes. Adv. Parasitol. 2016, 93, 69–94. [Google Scholar]
- Velan, A.; Hoda, M. In-silico comparison of inhibition of wild and drug-resistant Haemonchus contortus β-tubulin isotype-1 by glycyrrhetinic acid, thymol and albendazole interactions. J. Parasit. Dis. 2021, 45, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Costa-Junior, L.M.; Chaudhry, U.N.; Skuce, P.J.; Stack, S.; Sargison, N.D. A loop-mediated isothermal amplification (LAMP) assay to identify isotype 1 β-tubulin locus SNPs in synthetic double-stranded Haemonchus contortus DNA. J. Parasit. Dis. 2022, 46, 47–55. [Google Scholar] [CrossRef]
- Waseem, H.B.; Shakeel, M.; Hassan, F.-U.; Yaqoob, A.; Iqbal, A.; Khalid, A.; Akram, H.N.; Dilbar, N.; Qamar, S.; Tahir, R.A. In silico Identification and Computational Screening of Potential AFP Inhibitors Against Liver Cancer. Med. Chem. 2025, 21, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Kumar, P. From Data Science to Bioscience: Emerging era of bioinformatics applications, tools and challenges. Procedia Comput. Sci. 2023, 218, 1516–1528. [Google Scholar] [CrossRef]
- Sehgal, S.A.; Mannan, S.; Ali, S. Pharmacoinformatic and molecular docking studies reveal potential novel antidepressants against neurodegenerative disorders by targeting HSPB8. Drug Des. Dev. Ther. 2016, 10, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; Srinivas, K. Modern drug discovery process: An in silico approach. J. Bioinform. Seq. Anal. 2011, 2, 89–94. [Google Scholar]
- Sehgal, S.A.; Kanwal, S.; Tahir, R.A.; Khalid, Z.; Hammad, M.A. In silico elucidation of potential drug target sites of the Thumb Index Fold Protein, Wnt-8b. Trop. J. Pharm. Res. 2018, 17, 491–497. [Google Scholar] [CrossRef]
- Sehgal, S.A.; Hammad, M.A.; Tahir, R.A.; Akram, H.N.; Ahmad, F. Current therapeutic molecules and targets in neurodegenerative diseases based on in silico drug design. Curr. Neuropharmacol. 2018, 16, 649–663. [Google Scholar] [CrossRef]
- Tahir, R.A.; Sehgal, S.A.; Khattak, N.A.; Khan Khattak, J.Z.; Mir, A. Tumor necrosis factor receptor superfamily 10B (TNFRSF10B): An insight from structure modeling to virtual screening for designing drug against head and neck cancer. Theor. Biol. Med. Model. 2013, 10, 38. [Google Scholar] [CrossRef]
- Tur Razia, I.; Kanwal, A.; Riaz, H.F.; Malik, A.; Ahsan, M.; Saleem Khan, M.; Raza, A.; Sabir, S.; Sajid, Z.; Fardeen Khan, M. Recent trends in computer-aided drug design for anti-cancer drug discovery. Curr. Top. Med. Chem. 2023, 23, 2844–2862. [Google Scholar] [CrossRef]
- Agamah, F.E.; Mazandu, G.K.; Hassan, R.; Bope, C.D.; Thomford, N.E.; Ghansah, A.; Chimusa, E.R. Computational/in silico methods in drug target and lead prediction. Brief. Bioinform. 2020, 21, 1663–1675. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, M.R.; Wang, H.; Huang, D.; Wang, H.; Joseph, J.; Huang, T.; Zhang, F.; Wang, J.; Cheng, M. Discovery of small molecule inhibitors through pharmacophore modeling, molecular docking, molecular dynamics simulation and experimental validation against myeloid cell leukemia-1 (Mcl-1). J. Biomol. Struct. Dyn. 2021, 39, 2512–2525. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Tur Razia, I.; Kanwal, A.; Ahsan, M.; Tahir, R.A.; Sajid, M.; Khan, M.S.; Mukhtar, N.; Parveen, G.; Sehgal, S.A. Computational Advancement towards the Identification of Natural Inhibitors for Dengue Virus: A Brief Review. Comb. Chem. High Throughput Screen. 2024, 27, 2464–2484. [Google Scholar] [CrossRef]
- Kondapuram, S.K.; Sarvagalla, S.; Coumar, M.S. Docking-based virtual screening using PyRx Tool: Autophagy target Vps34 as a case study. In Molecular Docking for Computer-Aided Drug Design; Elsevier: Amsterdam, The Netherlands, 2021; pp. 463–477. [Google Scholar]
- Pawar, S.S.; Rohane, S.H. Review on discovery studio: An important tool for molecular docking. Asian J. Res. Chem. 2021, 14, 1–3. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 37. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32 (Suppl. 2), W526–W531. [Google Scholar] [CrossRef]
- Gopinath, P.; Kathiravan, M. Docking studies and molecular dynamics simulation of triazole benzene sulfonamide derivatives with human carbonic anhydrase IX inhibition activity. RSC Adv. 2021, 11, 38079–38093. [Google Scholar]
- Ovchinnikov, S.; Park, H.; Kim, D.E.; DiMaio, F.; Baker, D. Protein structure prediction using Rosetta in CASP12. Proteins Struct. Funct. Bioinform. 2018, 86, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Shivanika, C.; Kumar, D.; Ragunathan, V.; Tiwari, P.; Sumitha, A. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2022, 40, 585–611. [Google Scholar]
- Shoaib, T.H.; Abdelmoniem, N.; Mukhtar, R.M.; Alqhtani, A.T.; Alalawi, A.L.; Alawaji, R.; Althubyani, M.S.; Mohamed, S.G.; Mohamed, G.A.; Ibrahim, S.R. Molecular docking and molecular dynamics studies reveal the anticancer potential of medicinal-plant-derived lignans as MDM2-P53 interaction inhibitors. Molecules 2023, 28, 6665. [Google Scholar] [CrossRef]
- Azmi, M.B.; Sehgal, S.A.; Asif, U.; Musani, S.; Abedin, M.F.E.; Suri, A.; Ahmed, S.D.H.; Qureshi, S.A. Genetic insights into obesity: In silico identification of pathogenic SNPs in MBOAT4 gene and their structural molecular dynamics consequences. J. Biomol. Struct. Dyn. 2024, 42, 13074–13090. [Google Scholar] [CrossRef]
- Rizvi, S.M.D.; Hussain, T.; Mehmood, K.; Moin, A.; Alanazi, A.S.; Subaiea, G.M. Molecular docking and dynamic simulation study to explore quercetin as a multi-potent candidate against gliomas. Trop. J. Pharm. Res. 2021, 20, 815–823. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Abdallah, H.M.; Sindi, I.A.; Ibrahim, S.R.; Alzain, A.A. Unveiling the potential of phytochemicals to inhibit nuclear receptor binding SET domain protein 2 for cancer: Pharmacophore screening, molecular docking, ADME properties, and molecular dynamics simulation investigations. PLoS ONE 2024, 19, e0308913. [Google Scholar] [CrossRef]
- Das, N.R.; Sharma, T.; Goyal, N.; Singh, N.; Toropov, A.A.; Toropova, A.P.; Achary, P.G.R. Isoprenylcysteine carboxyl methyltransferase inhibitors: QSAR, docking and molecular dynamics studies. J. Mol. Struct. 2023, 1291, 135966. [Google Scholar] [CrossRef]
- Iqbal, A.; Waseem, H.B.; Ali, F.; Baammi, S.; Faheem, H.; Shazly, G.A.; Nafidi, H.-A.; Sajid, M.; Khan, M.S.; Akram, H.N. In silico identification and virtual screening to discover potent therapeutic phytochemicals against CMT2A. J. Indian Chem. Soc. 2024, 101, 101403. [Google Scholar] [CrossRef]
- Arsenopoulos, K.V.; Fthenakis, G.C.; Katsarou, E.I.; Papadopoulos, E. Haemonchosis: A challenging parasitic infection of sheep and goats. Animals 2021, 11, 363. [Google Scholar] [CrossRef]
- Ashraf, S.; Beech, R.N.; Hancock, M.A.; Prichard, R.K. Ivermectin binds to Haemonchus contortus tubulins and promotes stability of microtubules. Int. J. Parasitol. 2015, 45, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Luo, X.; Wang, R.; Zhai, B.; Wang, P.; Li, J.; Yang, X. Transcriptomics and proteomics of Haemonchus contortus in response to ivermectin treatment. Animals 2023, 13, 919. [Google Scholar] [CrossRef] [PubMed]
- Kotze, A.; Prichard, R. Anthelmintic resistance in Haemonchus contortus: History, mechanisms and diagnosis. Adv. Parasitol. 2016, 93, 397–428. [Google Scholar] [PubMed]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
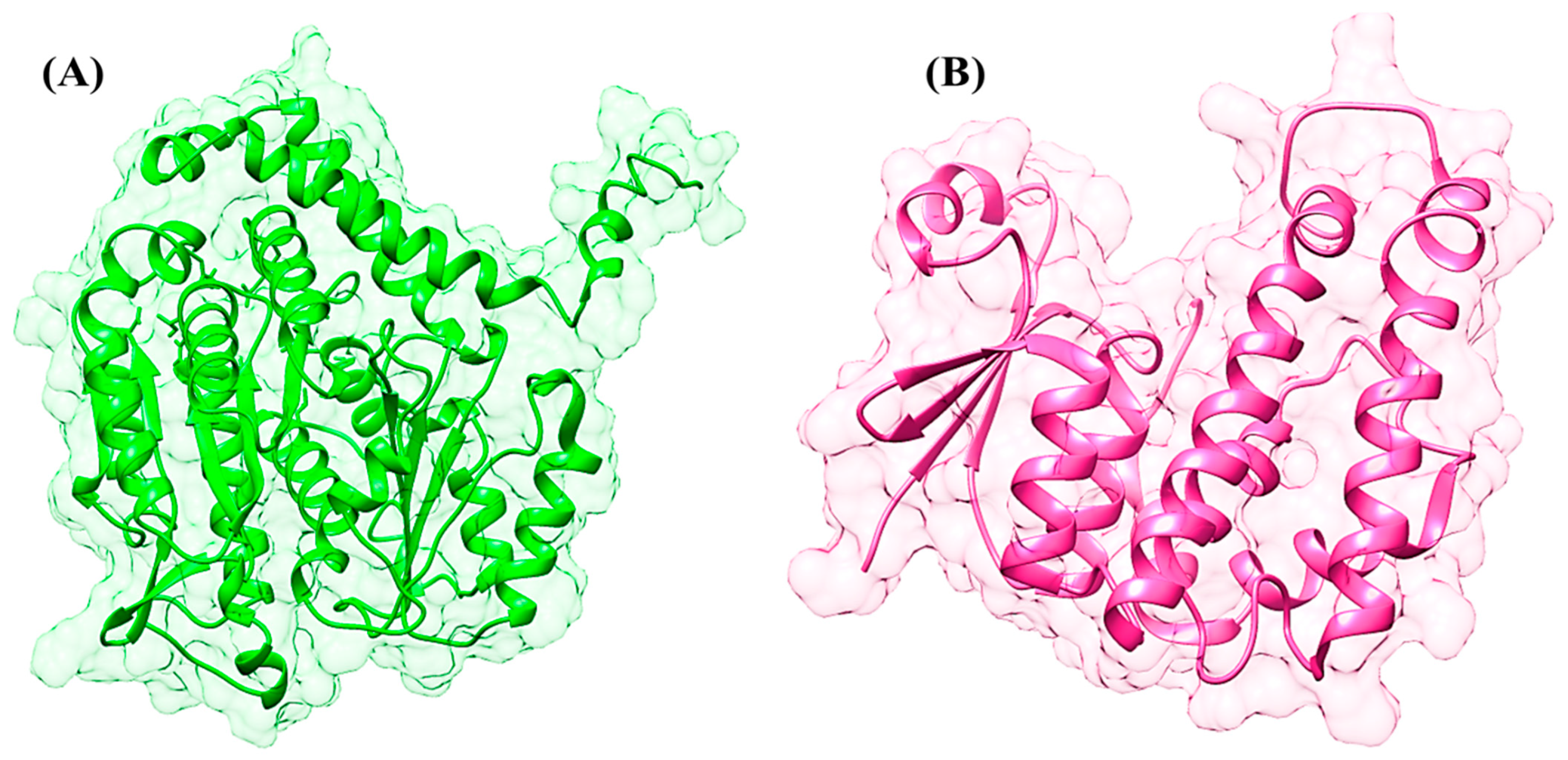

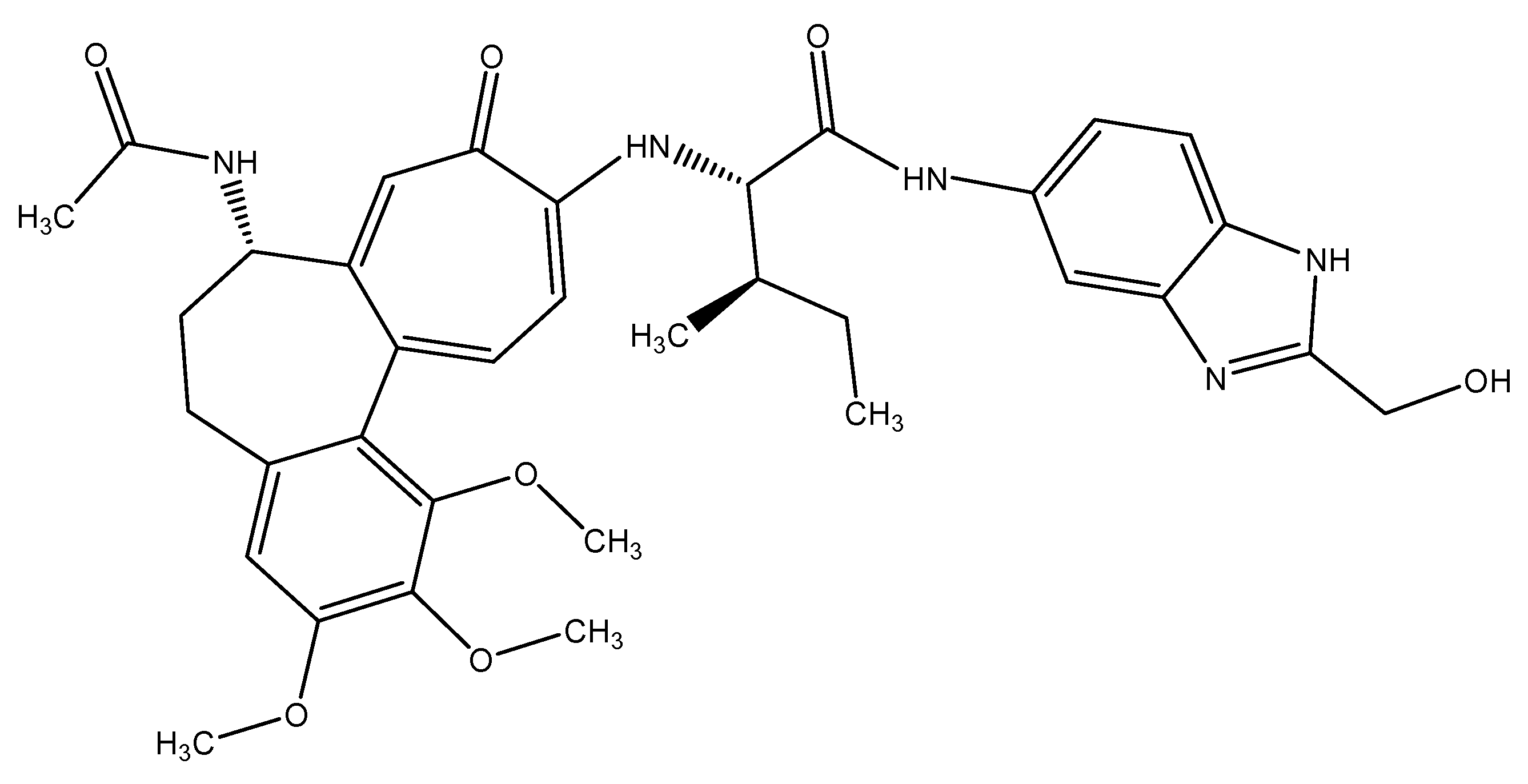



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds ID | With Beta-Tubulin Isotype-1 (Kcal/mol) | With GST (Kcal/mol) | Binding Residues (β-Tubulin Isotype-1) | Binding Residues (GST) |
|---|---|---|---|---|
| 9832750 | −8.7 | −10.4 | Arg2, Glu45, Arg46, Cys129, Gln131, Cys239, Phe242, Pro243, Gly-44, Asp249, Arg251, Lys252 | Tyr8, Gly13, Ala14, Gln63, Val65, Phe95, Lys96, Leu99, Asn100, Arg103, Phe106, Lys107, Glu162, Met163, Phe204 |
| 135449328 | −7.4 | −8.9 | Gly10, Gln11, Cys12, Gln131, Ser138, Gly144, Asp177, Thr178, Tyr222, Leu225 | Arg12, Gly13, Glu16, Ile17, Phe95, Leu99, Arg103, Phe106, Lys107, Leu110 Try159, Glu162, Met163, Thr166, His168, Ser202, Phe204 |
| 4030 | −7.8 | −8.2 | Gln11, Cys12, Asn99, Ser138, Leu139, Gly140, Gly141, Val178, Asp177, Glu181 | Arg12, Gly13, Ala14, Glu16, Ile17, Ser64, Phe95, Leu99, Arg103, Phe106, Lys107, Leu110 Try159, Glu162, Met163, Phe165, Thr166, His168, Lys203, Phe204 |
| 40854 | −7.5 | −7.8 | Cys12, Ala97, Asn99, Ser138, Leu139, Gly140, Gly141, Val169, Val175, Val178, Asp177 | Arg12, Gly13, Phe95, Arg103, Phe106, Lys107, Leu110 Try159, Glu162, Met163, Phe165, Thr166, His168, Lys203, Phe204 |
| 71449 | −6.7 | −7.8 | Gly10, Gln11, Cys12, Ala97, Asn99, Ser138, Leu139, Gly140, Gly141, Val169, Val175, Val178, Asp177, Asn204 | Arg12, Gly13, Ala14, Phe95, Leu99, Arg103, Phe106, Lys107, Leu110 Try159, Glu162, Met163, Phe165, Thr166, His168, Phe204 |
| 50248 | −6.6 | −7.5 | Gln11, Cys12, Asn99, Ser138, Leu139, Gly140, Gly141, Val178, Asp177, Glu181 | Arg12, Gly13, Ala14, Glu16, Ile17, Ser64, Phe95, Leu99, Arg103, Phe106, Lys107, Leu110 Try159, Glu162, Met163, Thr166, His168 |
| 83969 | −6.9 | −7.2 | Gly10, Gln11, Cys12, Glu69, Ala97, Gly98, Asn99, Ser138, Leu139, Gly140, Gly141, Val169, Val175, Val178, Asp177, Asn204 | Arg12, Gly13, Glu16, Ile17, Phe95, Leu99, Arg103, Phe106, Lys107, Leu110 Try159, Glu162, Met163, Phe165, Thr166, His168, Phe204 |
| 33309 | −7 | −7.1 | Cys12, Glu69, Thr72, Asn99, Ser138, Leu139, Gly140, Gly141, Val170, Pro171, Val175, Val178, Asp177, Asn204 | Arg12, Gly13, Ala14, Phe95, Leu99, Arg103, Phe106, Lys107, Try159, Glu162, Met163, Phe165, Thr166, Phe204 |
| 4622 | −6.3 | −7.1 | Gln11, Cys12, Ala97, Asn99, Ser138, Leu139, Gly140, Gly141, Val169, Val175, Val178, Asp177, Asn204 | Arg12, Gly13, Glu16, Ile17, Phe95, Leu99, Arg103, Phe106, Lys107, Leu110 Try159, Glu162, Met163, Phe165, Thr166, His168, Phe204 |
| 2082 | −6.3 | −7 | Gly10, Gln11, Cys12, Ala97, Asn99, Ser138, Leu139, Gly140, Gly141, Val169, Val175, Val178, Asp177 | Arg12, Gly13, Glu16, Ile17, Phe95, Leu99, Arg103, Phe106, Lys107, Leu110, Leu158, Try159, Glu162, Met163, Thr166 |
| 26879 | −5.8 | −6.9 | Gln11, Cys12, Ser138, Leu139, Gly140, Gly141, Val178, Asp177, Glu181 | Arg12, Gly13, Glu16, Ile17, Phe95, Try159, Glu162, Met163, Thr166 |
| 5430 | −6.2 | −6.7 | Gln11, Cys12, Asn99, Ser138, Leu139, Gly140, Gly141, Val169, Val175, Val178, Asp177, Asn204 | Arg12, Gly13, Glu16, Ile17, Phe95, Try159, Glu162, Met163, Thr166, Phe204 |
| Compounds | With β-Tubulin Isotype-1 (Kcal/mol) | With GST (Kcal/mol) |
|---|---|---|
| Molport-000-534-313 | −9.4 | −9.4 |
| Molport-039-195-358 | −9.4 | −9.5 |
| Molport-000-534-195 | −8.7 | −9.0 |
| Molport-000-534-076 | −10.2 | −8.4 |
| Molport-000-097-781 | −9.1 | −8.7 |
| Molport-000-534-254 | −8.9 | −8.7 |
| Molport-000-532-863 | −8.7 | −9.7 |
| Molport-000-092-797 | −8.8 | −8.8 |
| Molport-002-594-703 | −7.8 | −9.7 |
| Molport-000-532-878 | −8.8 | −7.9 |
| Model | Result | Probability |
|---|---|---|
| Adsorption | ||
| Blood–Brain Barrier | BBB− | 0.8433 |
| Human Intestinal Absorption | HIA+ | 0.9661 |
| Caco-2 Permeability | Caco-2 | 0.7124 |
| P-glycoprotein Substrate | Substrate | 0.7854 |
| P-glycoprotein Inhibitor | Non-inhibitor | 0.9112 |
| Distribution | ||
| Subcellular Localization | Nucleus | 0.4909 |
| Metabolism | ||
| CYP450 2C9 Substrate | Non-substrate | 0.7745 |
| CYP450 2D6 Substrate | Non-substrate | 0.8307 |
| CYP450 1A2 Inhibitor | Non-inhibitor | 0.6984 |
| CYP450 2C9 Inhibitor | Non-inhibitor | 0.6149 |
| CYP450 2D6 Inhibitor | Non-inhibitor | 0.8617 |
| CYP450 2C19 Inhibitor | Non-inhibitor | 0.7318 |
| CYP450 3A4 Inhibitor | Non-inhibitor | 0.7707 |
| Toxicity | ||
| AMES Toxicity | Non-AMES toxic | 0.6349 |
| Carcinogens | Non-carcinogens | 0.9172 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Sehgal, S.A.; Hassan, F.; Liu, G. In Silico Identification of Novel Compounds as Anthelmintics Against Haemonchus contortus Through Inhibiting β-Tubulin Isotype 1 and Glutathione S-Transferase. Animals 2025, 15, 1846. https://doi.org/10.3390/ani15131846
Jin Y, Sehgal SA, Hassan F, Liu G. In Silico Identification of Novel Compounds as Anthelmintics Against Haemonchus contortus Through Inhibiting β-Tubulin Isotype 1 and Glutathione S-Transferase. Animals. 2025; 15(13):1846. https://doi.org/10.3390/ani15131846
Chicago/Turabian StyleJin, Yaqian, Sheikh Arslan Sehgal, Faizul Hassan, and Guiqin Liu. 2025. "In Silico Identification of Novel Compounds as Anthelmintics Against Haemonchus contortus Through Inhibiting β-Tubulin Isotype 1 and Glutathione S-Transferase" Animals 15, no. 13: 1846. https://doi.org/10.3390/ani15131846
APA StyleJin, Y., Sehgal, S. A., Hassan, F., & Liu, G. (2025). In Silico Identification of Novel Compounds as Anthelmintics Against Haemonchus contortus Through Inhibiting β-Tubulin Isotype 1 and Glutathione S-Transferase. Animals, 15(13), 1846. https://doi.org/10.3390/ani15131846