
Mitogenome of Endemic Species of Flying Squirrel, Trogopterus xanthipes (Rodentia, Mammalia) and Phylogeny of the Sciuridae

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Collection and Identification
2.2. DNA Extraction, Mitogenome Sequencing and Analysis
2.3. Phylogenetic Analysis
3. Results
3.1. Morphological Characteristics of T. xanthipes
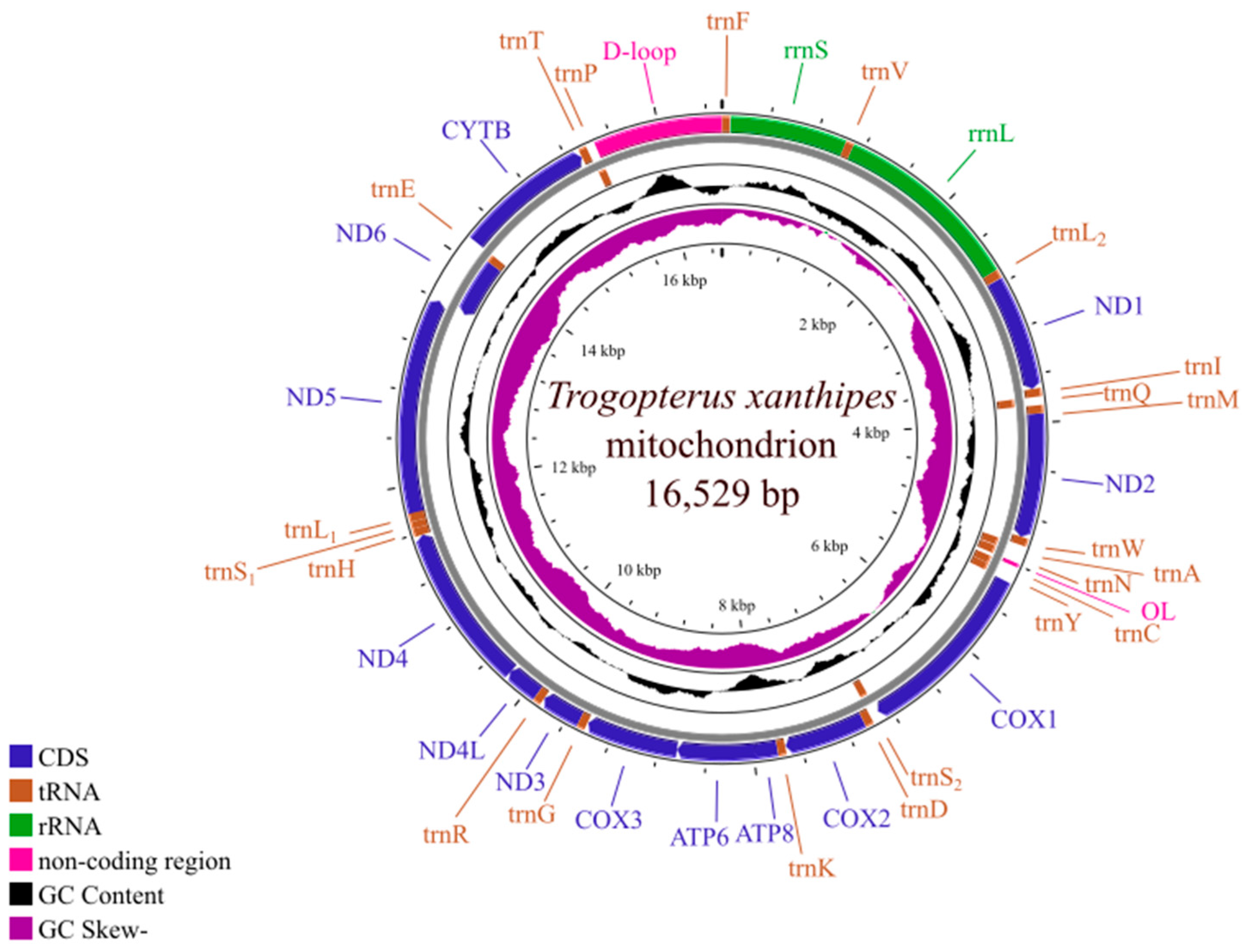
3.2. Mitogenome Organization of T. xanthipes
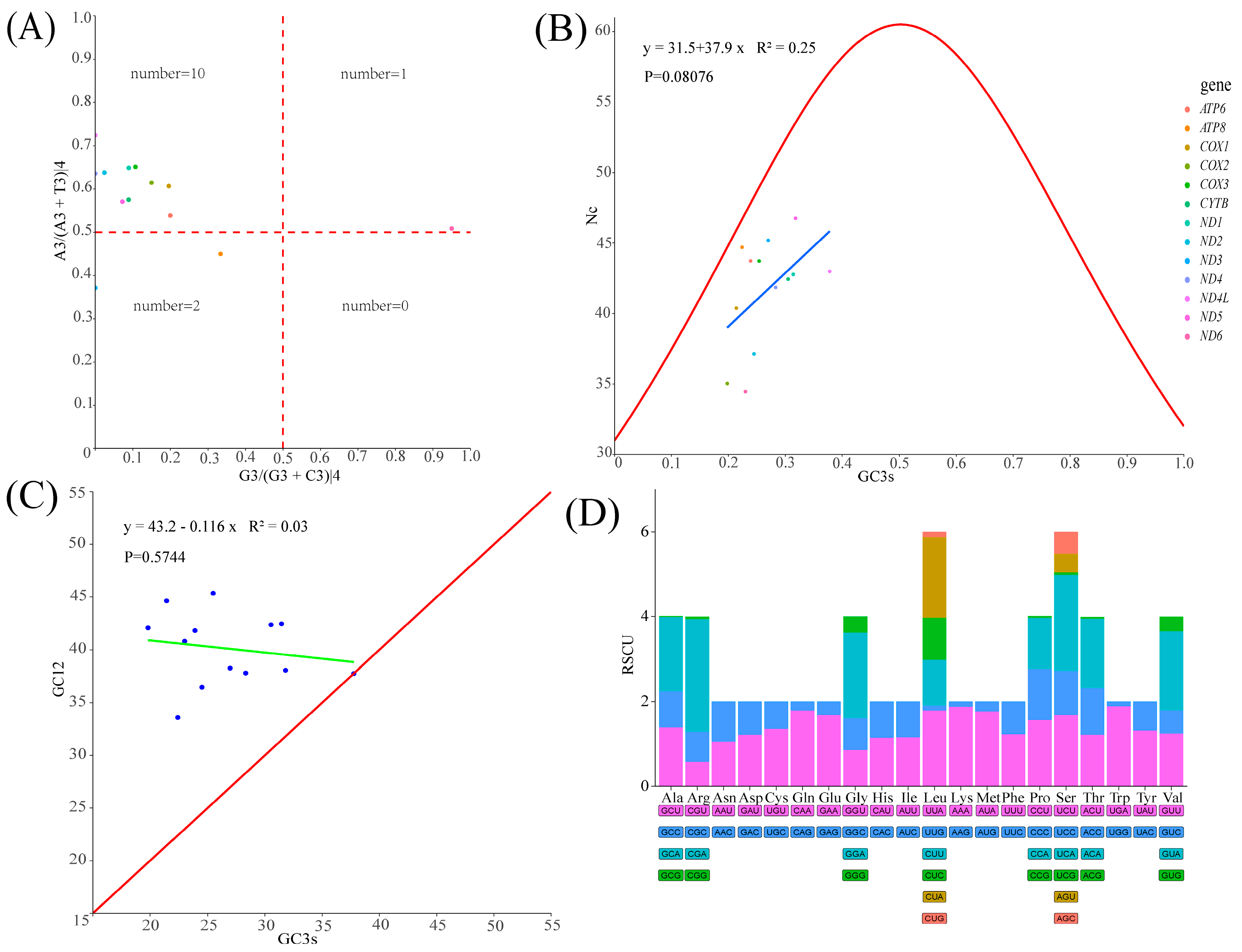
3.3. Protein-Coding Genes and Codon Usage Patterns
3.4. Ribosomal RNA and Transfer RNA Genes
3.5. Non-Coding Region
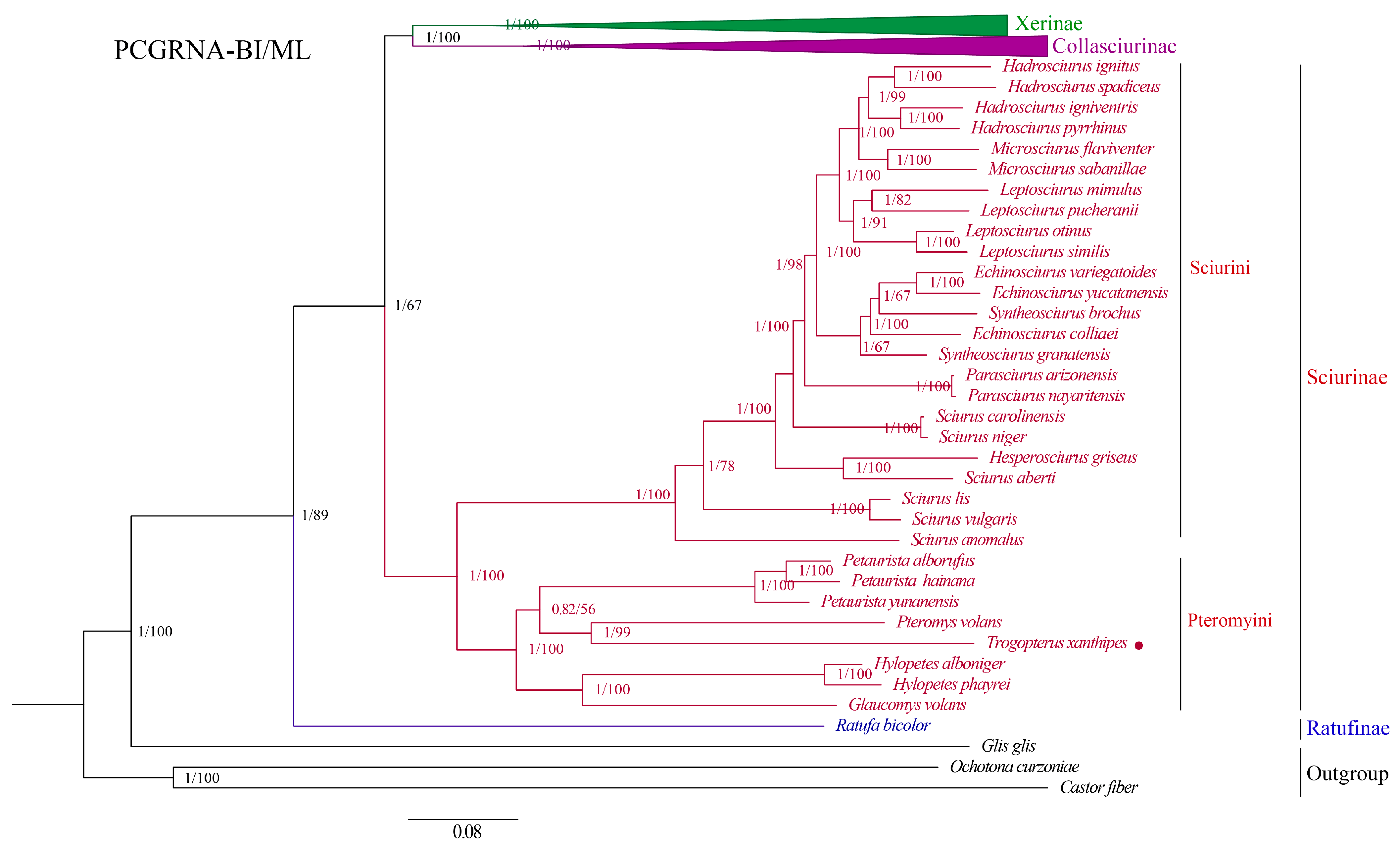
3.6. Phylogenetic Analysis
4. Discussion
4.1. Morphological Characteristics and Mitogenome of T. xanthipes
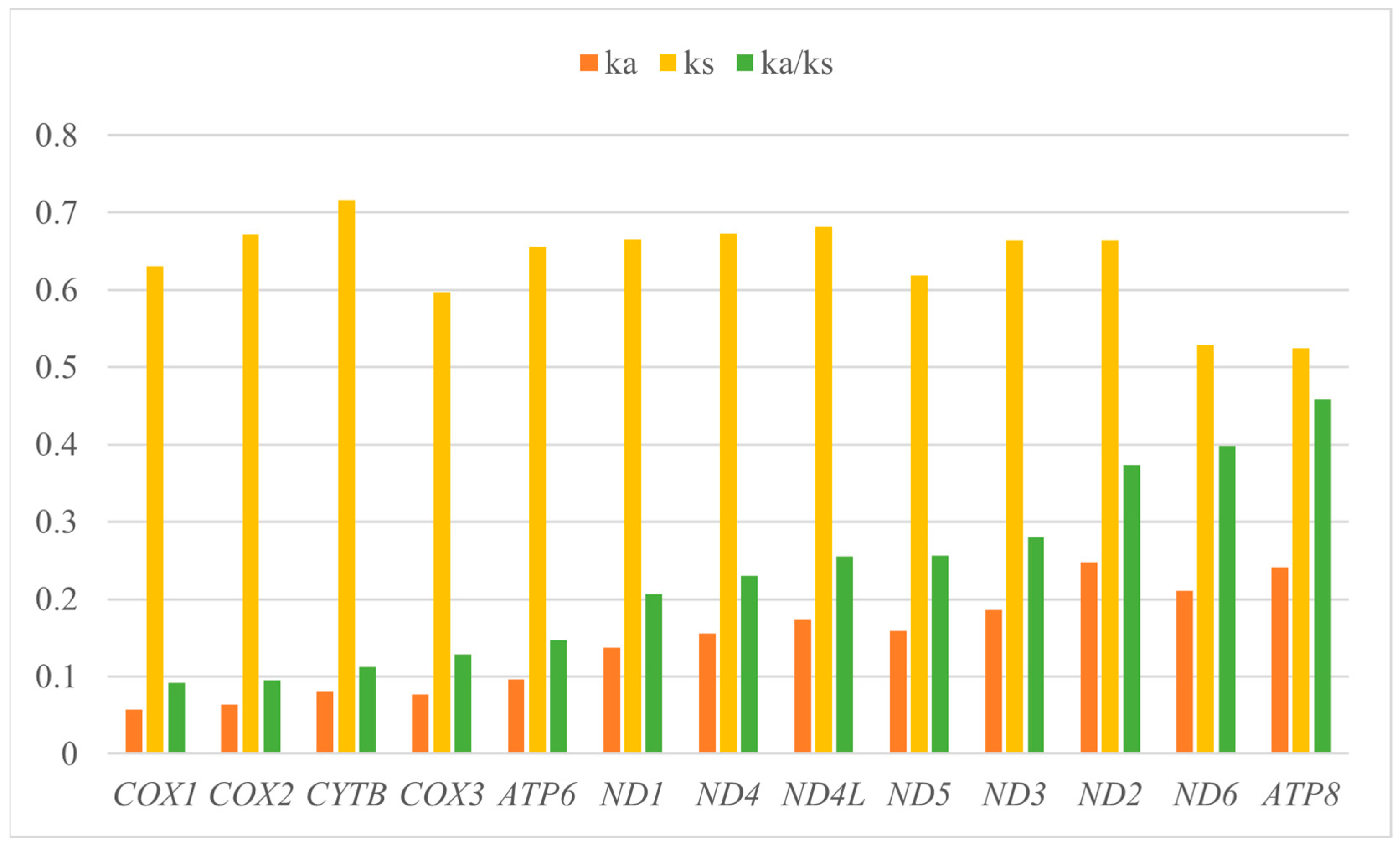
4.2. Comparative Mitogenomic Analysis of T. xanthipes and the Closest Related Species (P. volans)
4.3. Phylogeny of the Sciuridae Based on Mitogenomes Sequences
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, D.E.; Lacher, T.E., Jr.; Mittermeier, R.A. Hand Book of the Mammals of the Word; Lynx Edicions: Barcelona, Spain, 2016; Volume 6. [Google Scholar]
- Liu, S.Y.; Wu, Y.; Li, S. Hand Book of the Mammals of the China, 3rd ed.; Straits Book Company: Fuzhou, China, 2022. [Google Scholar]
- Andrew, T.; Smith, X.Y. A Guide to the Mammals of China; Hunan Education Press: Changsha, China, 2009. [Google Scholar]
- Pocock, R.I. The classification of the Sciuridae. Proc. Zool. Soc. Lond. 1923, 1, 209–246. [Google Scholar] [CrossRef]
- Bruijn, H.; Unay, E. Petauristinae (Mammalia, Rodentia) from the Oligocene of Spain, Belgium, and Turkish Thrace; Natural History Museum of Los Angeles County: Los Angeles, CA, USA, 1989. [Google Scholar]
- Ellerman, J.R. The Families and Genera of Living Rodents; Order of the Trustees of the British Museum: London, UK, 1940; Volume 1. [Google Scholar]
- Ellerman, J.R.; Morrison-Scott, T.C.S. Checklist of Palaearctic and Indian Mammals; British Museum (Natural History): London, UK, 1951. [Google Scholar]
- Hoffmann, R.S. Family Ochotonidae, 2nd ed.; Smithsonian Institution Press: Washington, WA, USA, 1993. [Google Scholar]
- Huang, W.J.; Chen, Y.H.; Wen, Y.X. Rodents in China; Fudan University Press: Shanghai, China, 1995. [Google Scholar]
- Wang, Y.X. Taxon and Complete Distribution of Mammal Species and Subspecies in China; China Forestry Press: Beijing, China, 2003. [Google Scholar]
- Mercer, J.M.; Roth, V.L. The effects of Cenozoic global change on squirrel phylogeny. Science 2003, 299, 1568–1572. [Google Scholar] [CrossRef]
- Steppan, S.J.; Storz, B.L.; Hoffmann, R.S. Nuclear DNA phylogeny of the squirrels (Mammalia: Rodentia) and the evolution of arboreality from c-myc and RAG1. Mol. Phylogenet. Evol. 2004, 30, 703–719. [Google Scholar] [CrossRef]
- Wilson, D.E.; Reeder, D.M. Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 2005. [Google Scholar]
- Wei, F.W. Taxonomy and Distribution of Mammals in China; Science Press: Beijing, China, 2022. [Google Scholar]
- Herron, M.D.; Castoe, T.A.; Parkinson, C.L. Sciurid phylogeny and the paraphyly of Holarctic ground squirrels (Spermophilus). Mol. Phylogenetics Evol. 2004, 31, 1015–1030. [Google Scholar] [CrossRef]
- Zhang, D.; Zou, H.; Wu, S.G.; Li, M.; Jakovlić, I.; Zhang, J.; Chen, R.; Wang, G.T.; Li, W.X. Sequencing of the complete mitochondrial genome of a fish-parasitic flatworm Paratetraonchoides inermis (Platyhelminthes: Monogenea): tRNA gene arrangement reshuffling and implications for phylogeny. Parasites Vectors 2017, 10, 462. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Shazwani Zakaria, S. Enhancing mitogenomic phylogeny and resolving the relationships of extinct megafaunal placental mammals. Mol. Phylogenetics Evol. 2021, 158, 107082. [Google Scholar] [CrossRef]
- Thalmann, O.; Shapiro, B.; Cui, P.; Schuenemann, V.J.; Sawyer, S.K.; Greenfield, D.L.; Germonpré, M.B.; Sablin, M.V.; López-Giráldez, F.; Domingo-Roura, X.; et al. Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs. Science 2013, 342, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Li, W.; Liao, J. Characterization of the complete mitochondrial genome of Phodopus roborovskii (Rodentia: Cricetidae) and systematic implications for Cricetinae phylogenetics. Biochem. Syst. Ecol. 2016, 69, 226–235. [Google Scholar] [CrossRef]
- Ding, L.; Li, W.; Liao, J. Mitochondrial genome of Cricetulus migratorius (Rodentia: Cricetidae): Insights into the characteristics of the mitochondrial genome and the phylogenetic relationships of Cricetulus species. Gene 2016, 595, 121–129. [Google Scholar] [CrossRef]
- Frank, K.; Barta, E.; Bana, N.Á.; Nagy, J.; Horn, P.; Orosz, L.; Stéger, V. Complete mitochondrial genome sequence of a Hungarian red deer (Cervus elaphus hippelaphus) from high-throughput sequencing data and its phylogenetic position within the family Cervidae. Acta Biol. Hung. 2016, 67, 133–147. [Google Scholar] [CrossRef]
- Sarvani, R.K.; Parmar, D.R.; Tabasum, W.; Thota, N.; Sreenivas, A.; Gaur, A. Characterization of the complete mitogenome of Indian Mouse Deer, Moschiola indica (Artiodactyla: Tragulidae) and its evolutionary significance. Sci. Rep. 2018, 8, 2697. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.D.; Song, H.; Cameron, S.L.; Whiting, M.F. A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol. Phylogenet. Evol. 2008, 49, 59–68. [Google Scholar] [CrossRef]
- Jacobsen, M.W.; Hansen, M.M.; Orlando, L.; Bekkevold, D.; Bernatchez, L.; Willerslev, E.; Gilbert, M.T.P. Mitogenome sequencing reveals shallow evolutionary histories and recent divergence time between morphologically and ecologically distinct European whitefish (Coregonus spp.). Mol. Ecol. 2012, 21, 2727–2742. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, S.; Paupério, J.; Pavlova, S.V.; Alves, P.C.; Searle, J.B. The Microtus voles: Resolving the phylogeny of one of the most speciose mammalian genera using genomics. Mol. Phylogenet. Evol. 2018, 125, 85–92. [Google Scholar] [CrossRef]
- Xu, X.; Janke, A.; Arnason, U. The complete mitochondrial DNA sequence of the greater Indian rhinoceros, Rhinoceros unicornis, and the phylogenetic relationship among Carnivora, Perissodactyla, and Artiodactyla (+Cetacea). Mol. Biol. Evol 1996, 13, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Waddell, P.J.; Cao, Y.; Hauf, J.; Hasegawa, M. Using novel phylogenetic methods to evaluate mammalian mtDNA, including amino acid-invariant sites-LogDet plus site stripping, to detect internal conflicts in the data, with special reference to the positions of hedgehog, armadillo and elephant. Syst. Biol. 1999, 48, 31–53. [Google Scholar] [CrossRef]
- Boukhdoud, L.; Parker, L.D.; McInerney, N.R.; Saliba, C.; Kahale, R.; Cross, H.; Matisoo-Smith, E.; Maldonado, J.E.; Bou Dagher Kharrat, M. First mitochondrial genome of the Caucasian squirrel Sciurus anomalus (Rodentia, Sciuridae). Mitochondrial DNA Part B 2021, 6, 883–885. [Google Scholar] [CrossRef]
- İbiş, O.; Selçuk, A.Y.; Teber, S.; Baran, M.; Koepfli, K.P.; Kefelioğlu, H.; Tez, C. Mitogenomic Analysis of Glirids (Gliridae) and Squirrels (Sciuridae) From Türkiye: Evolutionary and Taxonomic Implications Within the Suborder Sciuromorpha. Ecol. Evol. 2025, 15, e70956. [Google Scholar] [CrossRef]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Predict; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S.; Battistuzzi, F.U. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Du, W.; Sun, Q.; Hu, S.; Yu, P.; Kan, S.; Zhang, W. Equus mitochondrial pangenome reveals independent domestication imprints in donkeys and horses. Sci. Rep. 2025, 15, 6803. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Xiang, C.Y.; Gao, F.; Jakovlić, I.; Lei, H.P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Kazutaka Katoh, K.M. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Ranwez, V.; Harispe, S.; Delsuc, F.; Douzery, E.J.P. MACSE: Multiple Alignment of Coding SEquences Accounting for Frameshifts and Stop Codons. PLoS ONE 2011, 6, e22594. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J.; Kjer, K.; Page, R.; Sullivan, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Cong, H.Y.; Kong, L.M.; Motokawa, M.; Wang, W.Q.; Li, Y.C. Complete mitochondrial genome of maritime striped squirrel Tamiops maritimus (Rodentia: Sciuridae). Mitochondrial DNA Part A 2015, 28, 240–241. [Google Scholar] [CrossRef]
- İbiş, O.; Selçuk, A.Y.; Teber, S.; Baran, M.; Kaya, A.; Özcan, S.; Kefelioğlu, H.; Tez, C. Complete mitogenomes of Turkish tree squirrels, Sciurus anomalus and S. vulgaris, (Sciuridae: Rodentia: Mammalia) and their phylogenetic status within the tribe Sciurini. Gene 2022, 841, 146773. [Google Scholar] [CrossRef]
- Matrosova, V.A.; Gündüz, İ.; Ermakov, O.A.; Demirtaş, S.; Simonov, E. The complete mitochondrial genome of endemic Taurus ground squirrel Spermophilus taurensis (Rodentia: Sciuridae). Russ. J. Theriol. 2023, 22, 97–101. [Google Scholar] [CrossRef]
- Zhao, Y.; Liang, J.; Li, J.; Zhang, Z.; Sun, Y.; Liu, F.; Zhang, X.; Liang, Y.; Teng, L.; Liu, Z. First complete mitochondrial genome of the Alashan ground squirrel (Spermophilus alashanicus) (Rodentia: Sciuridae) from Ningxia, China. Mitochondrial DNA Part B 2024, 9, 148–152. [Google Scholar] [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J.; Collins, T. Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Tong, L. Mitochondrial poly(A) polymerase and polyadenylation. Biochim. Biophys. Acta (BBA)–Gene Regul. Mech. 2012, 1819, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Lal, D.; Verma, M.; Behura, S.K.; Lal, R. Codon usage bias in phylum Actinobacteria: Relevance to environmental adaptation and host pathogenicity. Res. Microbiol. 2016, 167, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The effective number of codons used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Liu, J.B.; Zhu, D.; Ma, G.; Liu, M.; Wang, M.; Jia, R.; Chen, S.; Sun, K.; Yang, Q.; Wu, Y.; et al. Genome-Wide Analysis of the Synonymous Codon Usage Patterns in Riemerella anatipestifer. Int. J. Mol. Sci. 2016, 17, 1304. [Google Scholar] [CrossRef]
- Tu, F.Y.; Qiao, Y.Q.; Zhao, W.J.; Wu, T. Comparative selective pressure analysis on mitochondrial protein-coding genes in flying squirrels (Pteromyini) and tree squirrels (Sciurini). Mitochondrial DNA Part A 2024, 35, 75–83. [Google Scholar] [CrossRef]
- Steinke, D.; Clare, E.L.; Lim, B.K.; Fenton, M.B.; Hebert, P.D.N. Neotropical Bats: Estimating Species Diversity with DNA Barcodes. PLoS ONE 2011, 6, e22648. [Google Scholar] [CrossRef]
- Strazewski, P.; Biala, E.; Gabriel, K.; McClain, W.H. The relationship of thermodynamic stability at a G × U recognition site to tRNA aminoacylation specificity. RNA 1999, 5, 1490–1494. [Google Scholar] [CrossRef]
- Wu, D.; Zhou, L.Z.; Xue, J.Z.; Xia, Q.L.; Meng, L. Characterization of Two New Apodemus Mitogenomes (Rodentia: Muridae) and Mitochondrial Phylogeny of Muridae. Diversity 2022, 14, 1089. [Google Scholar] [CrossRef]
- Chang, Y.B.; Yang, Q.S.; Wen, Z.X. Population genetic structure and demographic history of Ochotona cansus (Lagomorpha, Ochotonidae). Acta Theriol. Sin. 2016, 36, 373. [Google Scholar] [CrossRef]
- Ding, L.; Zhou, Q.; Sun, Y.H.; Feoktistova, N.Y.; Liao, J.C. Two novel cricetine mitogenomes: Insight into the mitogenomic characteristics and phylogeny in Cricetinae (Rodentia: Cricetidae). Genomics 2020, 112, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Luo, G.J.; Zhou, Q.; Sun, Y.H.; Liao, J.C. Comparative Mitogenome Analysis of Gerbils and the Mitogenome Phylogeny of Gerbillinae (Rodentia: Muridae). Biochem. Genet. 2022, 60, 2226–2249. [Google Scholar] [CrossRef]
- Cong, H.Y. Phylogenetic Analysis of Sciuridae Based on Mitochondrial Whole Genome and Expression Evaluation of Different Genes; Shandong University: Shandong, China, 2016. [Google Scholar]
- Lu, X.; Ge, D.; Xia, L.; Zhang, Z.; Li, S.; Yang, Q. The Evolution and Paleobiogeography of Flying Squirrels (Sciuridae, Pteromyini) in Response to Global Environmental Change. Evol. Biol. 2012, 40, 117–132. [Google Scholar] [CrossRef]
- Yu, F.R.; Yu, F.; Pang, J.; Kilpatrick, C.W.; McGuire, P.M.; Wang, Y.; Lu, S.; Woods, C.A. Phylogeny and biogeography of the Petaurista philippensis complex (Rodentia: Sciuridae), inter- and intraspecific relationships inferred from molecular and morphometric analysis. Mol. Phylogenet. Evol. 2006, 38, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Li, G.G.; Lwin, Y.H.; Yang, B.; Qin, T.; Phothisath, P.; Maung, K.-W.; Quan, R.-C.; Li, S. Taxonomic revision and phylogenetic position of the flying squirrel genus Biswamoyopterus (Mammalia, Rodentia, Sciuridae, Pteromyini) on the northern Indo-China peninsula. ZooKeys 2020, 939, 65–85. [Google Scholar] [CrossRef]
- Zelditch, M.L.; Li, J.C.; Tran, L.A.P.; Swiderski, D.L. Relationships of diversity, disparity, and their evolutionary rates in squirrels (Sciuridae). Evolution 2015, 69, 1284–1300. [Google Scholar] [CrossRef]
- Hinckley, A.; Hawkins, M.T.R.; Achmadi, A.S.; Maldonado, J.E.; Leonard, J.A. Ancient Divergence Driven by Geographic Isolation and Ecological Adaptation in Forest Dependent Sundaland Tree Squirrels. Front. Ecol. Evol. 2020, 8, 208. [Google Scholar] [CrossRef]
- Casanovas-Vilar, I.; Garcia-Porta, J.; Fortuny, J.; Sanisidro, Ó.; Prieto, J.; Querejeta, M.; Llácer, S.; Robles, J.M.; Bernardini, F.; Alba, D.M. Oldest skeleton of a fossil flying squirrel casts new light on the phylogeny of the group. eLife 2018, 7, e39270. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Wang, Z.; Song, W.; Dong, W. Mitogenome of Endemic Species of Flying Squirrel, Trogopterus xanthipes (Rodentia, Mammalia) and Phylogeny of the Sciuridae. Animals 2025, 15, 1493. https://doi.org/10.3390/ani15101493
Zhao D, Wang Z, Song W, Dong W. Mitogenome of Endemic Species of Flying Squirrel, Trogopterus xanthipes (Rodentia, Mammalia) and Phylogeny of the Sciuridae. Animals. 2025; 15(10):1493. https://doi.org/10.3390/ani15101493
Chicago/Turabian StyleZhao, Di, Zhongsong Wang, Wenyu Song, and Wenge Dong. 2025. "Mitogenome of Endemic Species of Flying Squirrel, Trogopterus xanthipes (Rodentia, Mammalia) and Phylogeny of the Sciuridae" Animals 15, no. 10: 1493. https://doi.org/10.3390/ani15101493
APA StyleZhao, D., Wang, Z., Song, W., & Dong, W. (2025). Mitogenome of Endemic Species of Flying Squirrel, Trogopterus xanthipes (Rodentia, Mammalia) and Phylogeny of the Sciuridae. Animals, 15(10), 1493. https://doi.org/10.3390/ani15101493