
Metagenomic Comparison of Gut Microbes of Lemur catta in Captive and Semi-Free-Range Environments



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Housing Conditions
2.2. Dietary Management
2.3. Sample Selection Criteria and Sample Collection
2.4. DNA Extraction and Metagenomic Sequencing
2.5. Bioinformatic Analysis
2.6. Statistical Analysis
3. Results
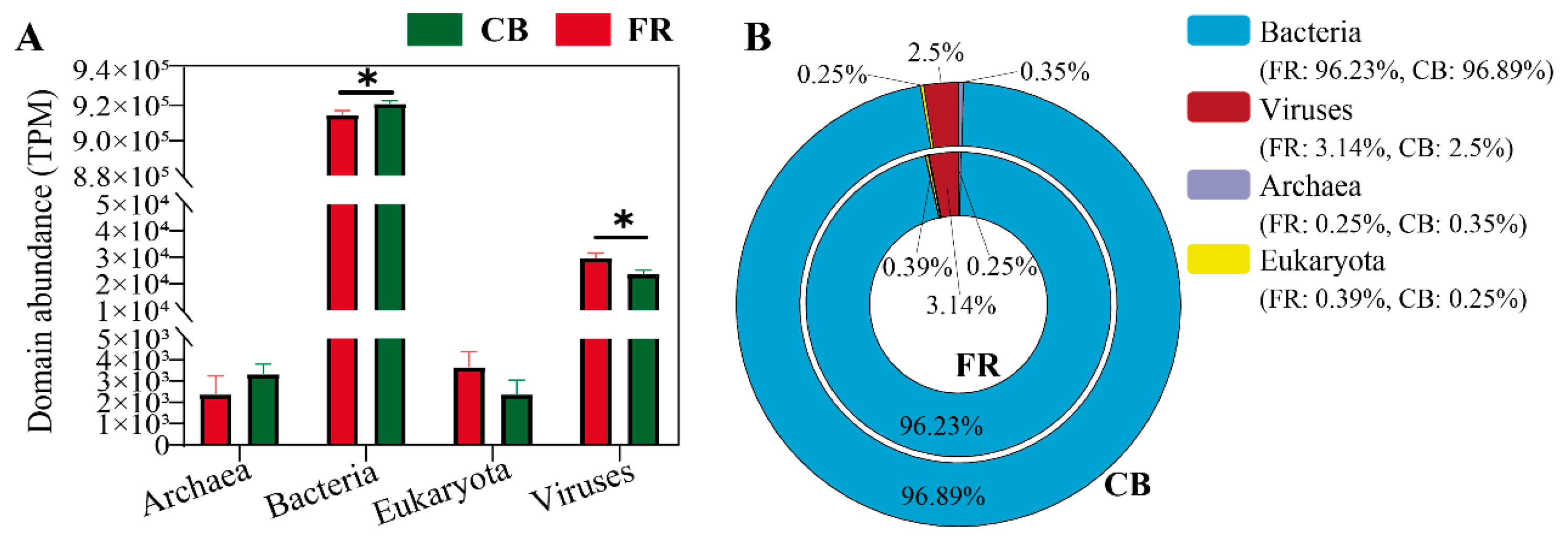
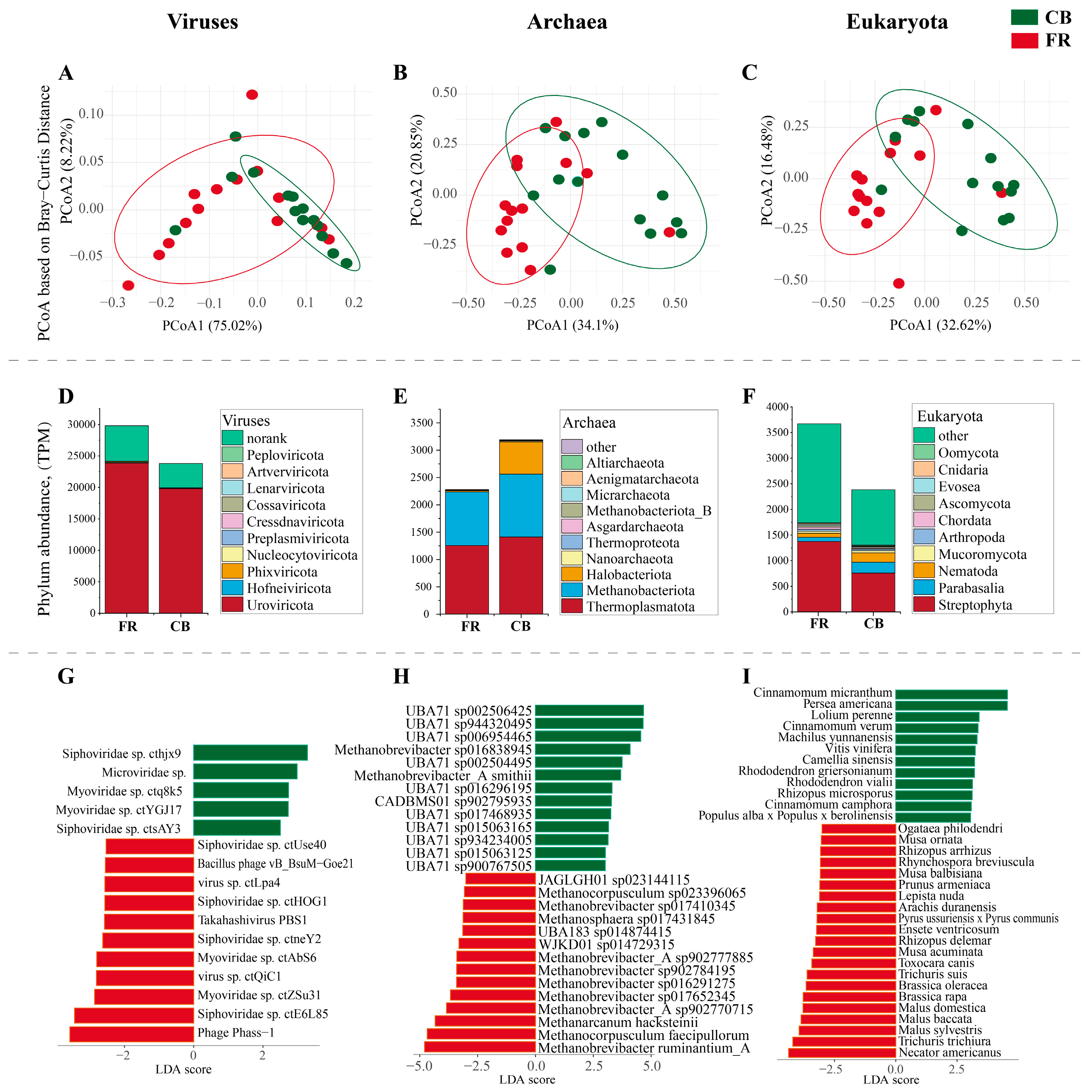
3.1. The Fecal Microbiota Composition at Domain Level and Viral Level
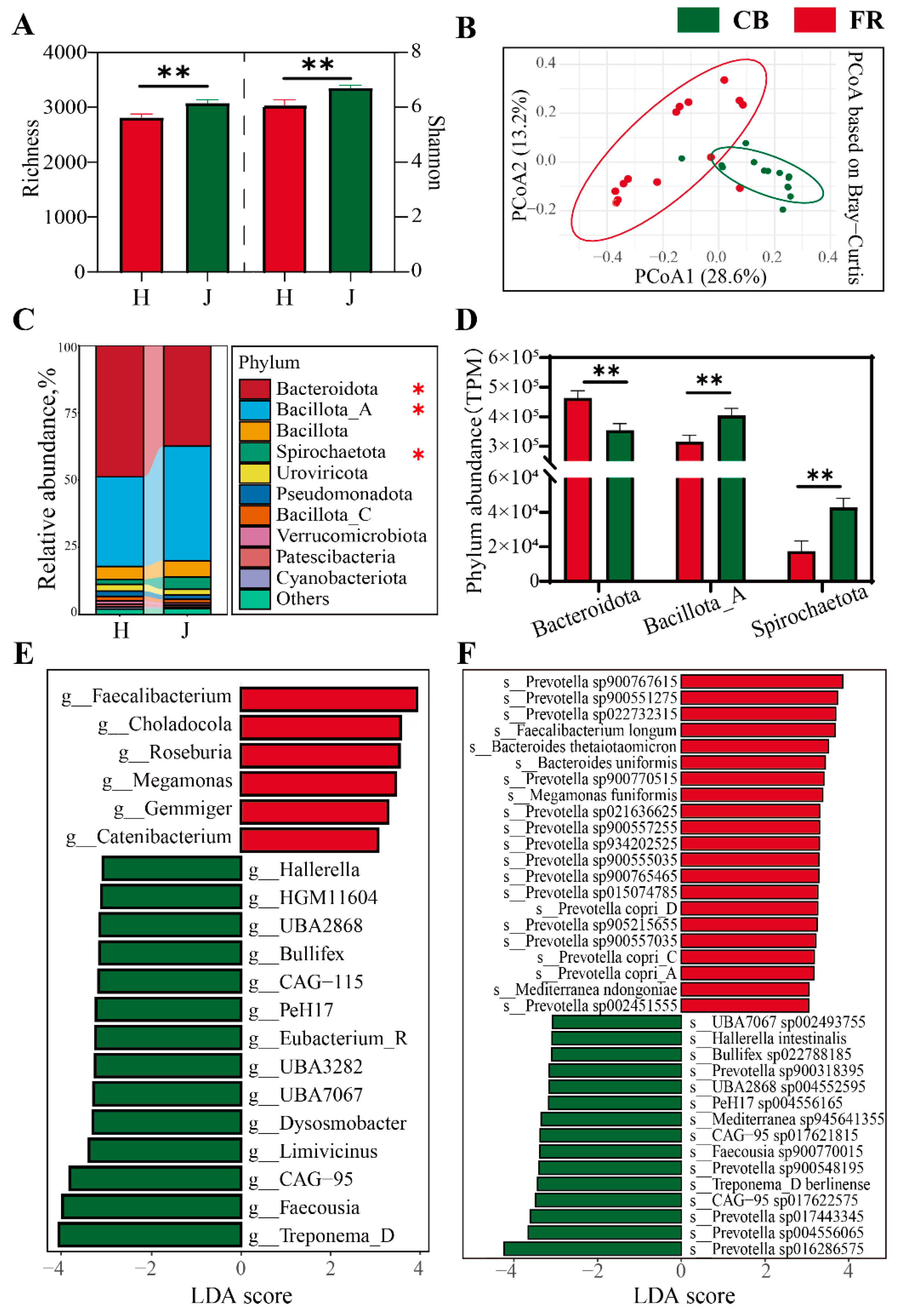
3.2. Analysis of Fecal Microbial Diversity and Composition at Bacterial Level
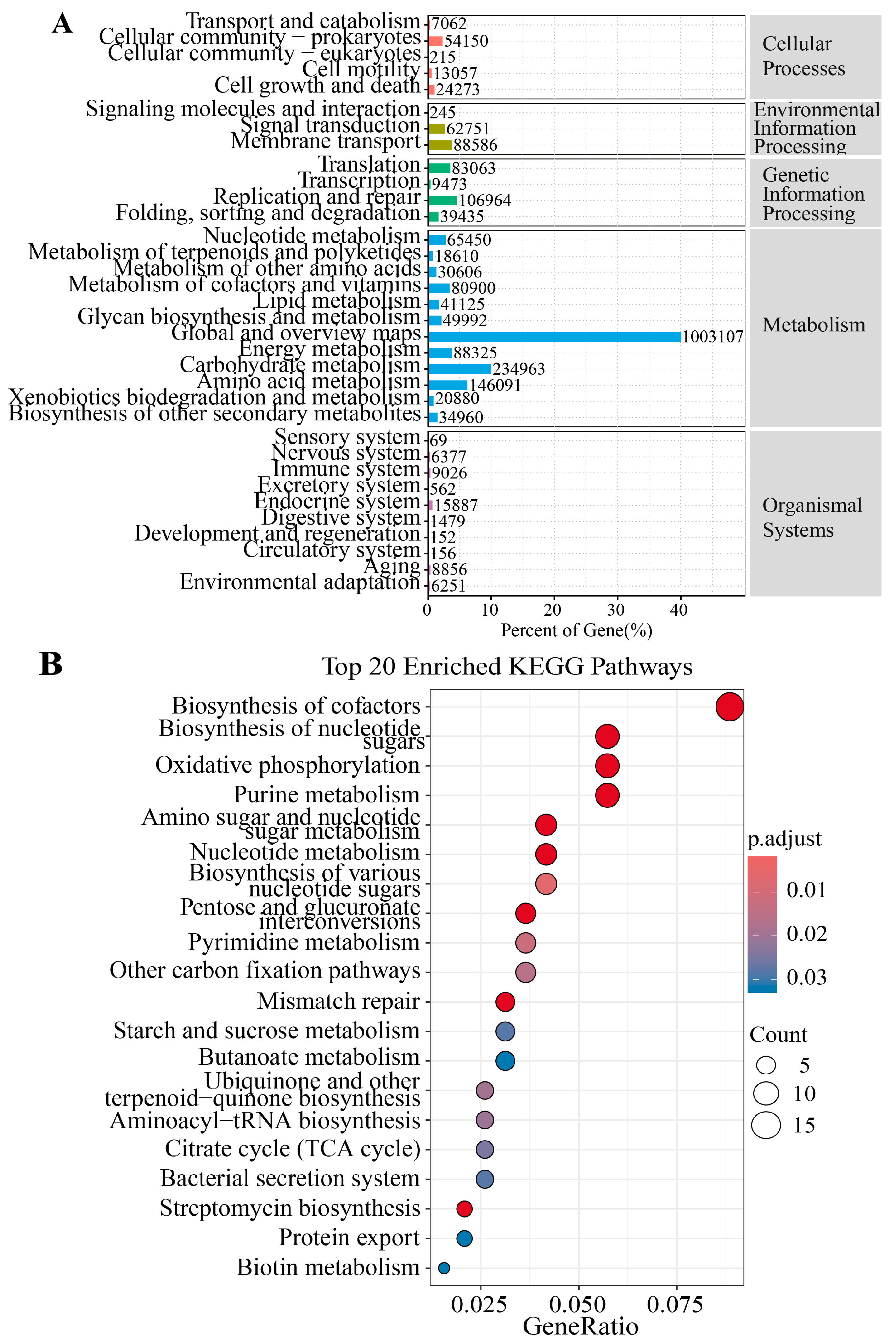
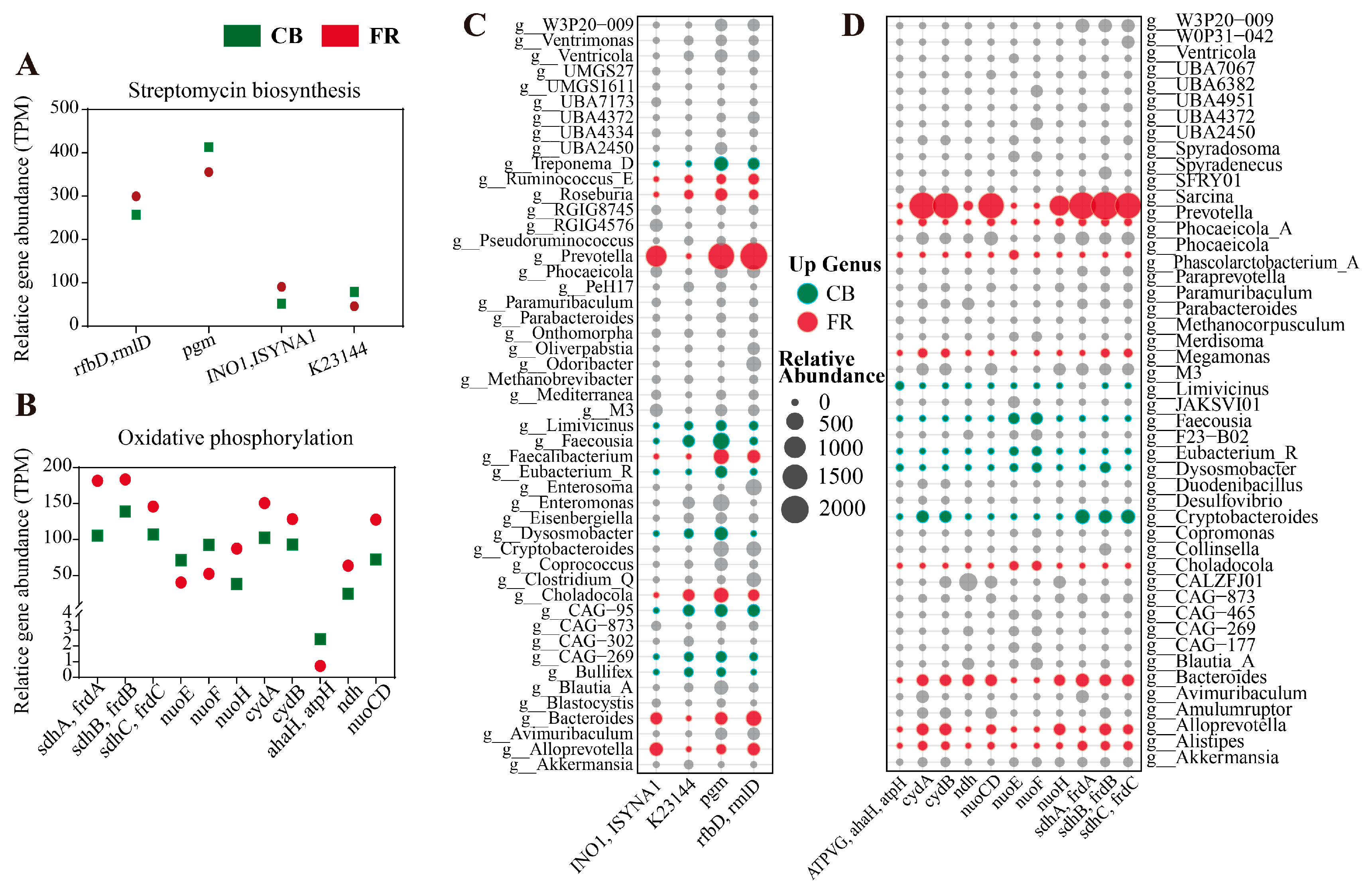
3.3. Metagenomic Analysis of the Differences in Metabolic Pathways
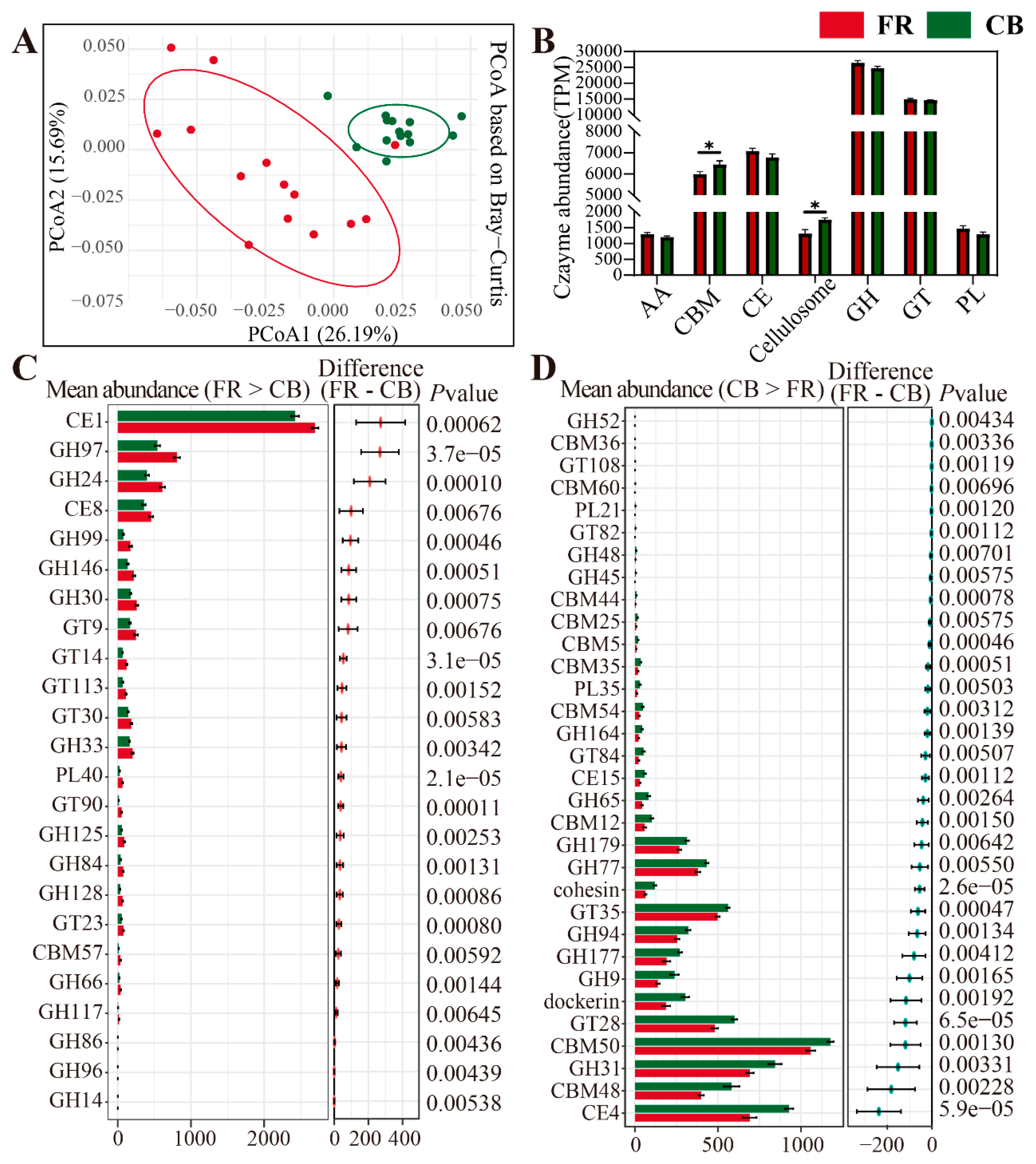
3.4. Metagenomic Analysis of the Differentially Represented Genes Encoding CAZymes
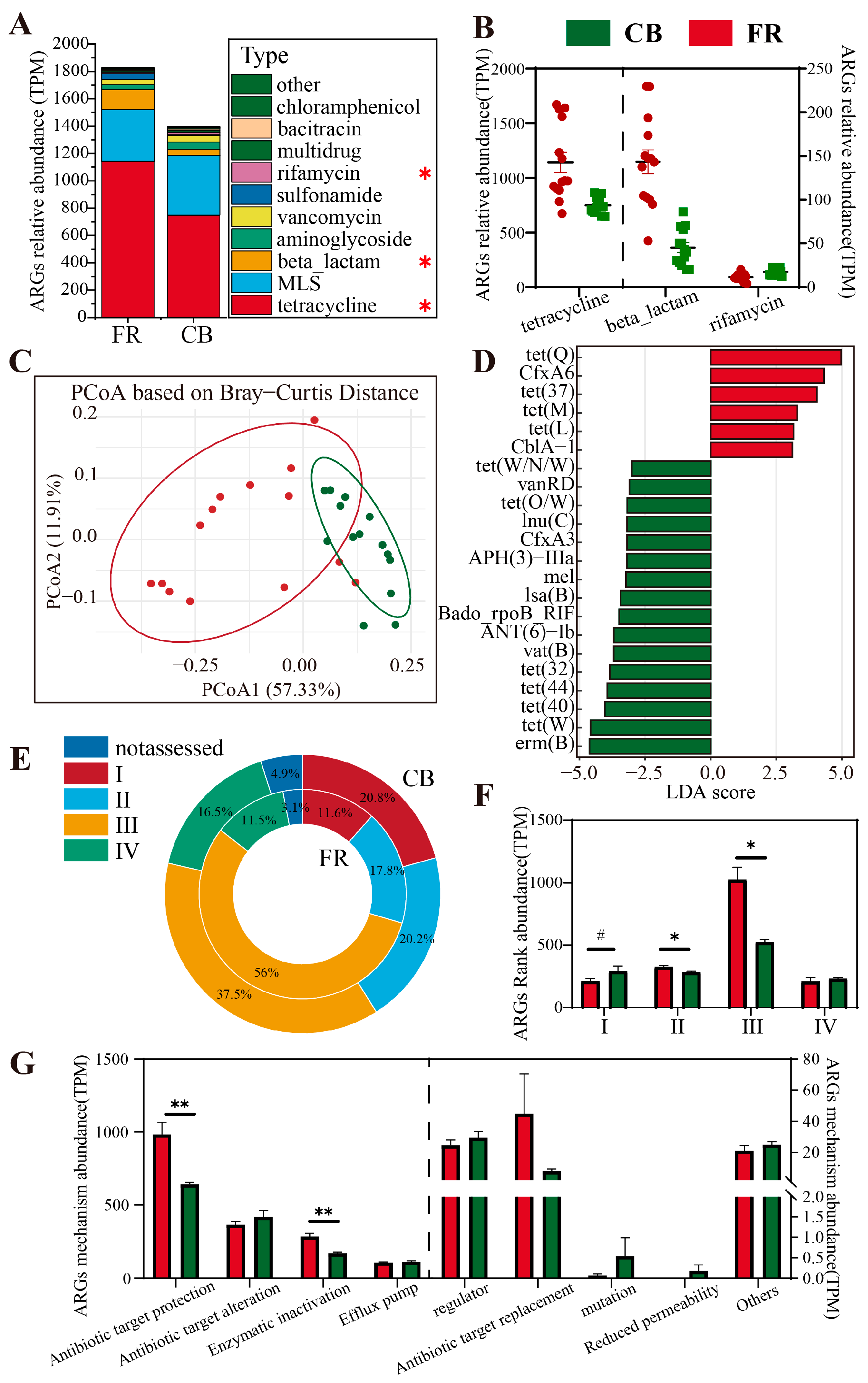
3.5. Metagenomic Analysis of Antimicrobial Resistance Genes (ARGs)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sheflin, A.M.; Melby, C.L.; Carbonero, F.; Weir, T.L. Linking dietary patterns with gut microbial composition and function. Gut Microbes 2017, 8, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Yang, X.; Li, G.; Zhu, Y.; Zhang, Z. Habitats Show More Impacts Than Host Species in Shaping Gut Microbiota of Sympatric Rodent Species in a Fragmented Forest. Front. Microbiol. 2022, 13, 811990. [Google Scholar] [CrossRef] [PubMed]
- Sonnega, S.; Sheriff, M.J. Harnessing the gut microbiome: A potential biomarker for wild animal welfare. Front. Vet. Sci. 2024, 11, 1474028. [Google Scholar] [CrossRef]
- Bennett, G.; Malone, M.; Sauther, M.L.; Cuozzo, F.P.; White, B.; Nelson, K.E.; Stumpf, R.M.; Knight, R.; Leigh, S.R.; Amato, K.R. Host age, social group, and habitat type influence the gut microbiota of wild ring-tailed lemurs (Lemur catta). Am. J. Primatol. 2016, 78, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Singleton, C.L.; Norris, A.M.; Sauther, M.L.; Cuozzo, F.P.; Jacky, I.A.Y. Ring-Tailed Lemur (Lemur catta) Health Parameters across Two Habitats with Varied Levels of Human Disturbance at the Bezà Mahafaly Special Reserve, Madagascar. Folia Primatol. 2015, 86, 56–65. [Google Scholar] [CrossRef]
- Schmidt, E.; Mykytczuk, N.; Schulte-Hostedde, A. Effects of the captive and wild environment on diversity of the gut microbiome of deer mice (Peromyscus maniculatus). ISME J. 2019, 13, 1293–1305. [Google Scholar] [CrossRef]
- Caselli, M.; Messeri, P.; Dessì-Fulgheri, F.; Bandoli, F. Enriching Zoo-Housed Ring-Tailed Lemurs (Lemur catta): Assessing the Influence of Three Types of Environmental Enrichment on Behavior. Animals 2022, 12, 2836. [Google Scholar] [CrossRef]
- Sauther, M.L.; Gould, L.; Cuozzo, F.P.; O’mara, M.T. Ring-tailed lemurs: A species re-imagined. Folia Primatol. 2015, 86, 5–13. [Google Scholar] [CrossRef]
- Bornbusch, S.L.; Clarke, T.A.; Hobilalaina, S.; Reseva, H.S.; LaFleur, M.; Drea, C.M. Microbial rewilding in the gut microbiomes of captive ring-tailed lemurs (Lemur catta) in Madagascar. Sci. Rep. 2022, 12, 22388. [Google Scholar] [CrossRef]
- Goodman, S.; Rakotoarisoa, S.V.; Wilmé, L. The distribution and biogeography of the ring-tailed lemur (Lemur catta). In Ring-Tailed Lemur Biology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 3–15. [Google Scholar]
- Schwitzer, C.; King, T.; Robsomanitrandrasana, E.; Chamberlan, C.; Rasolofoharivelo, T. Integrating ex situ and in situ conservation of lemurs. In Lemurs of Madagascar: A Strategy for their Conservation 2013–2016; IUCN SSC Primate Specialist Group: Austin, TX, USA, 2013; pp. 146–152. [Google Scholar]
- Bornbusch, S.L.; Greene, L.K.; Rahobilalaina, S.; Calkins, S.; Rothman, R.S.; Clarke, T.A.; LaFleur, M.; Drea, C.M. Gut microbiota of ring-tailed lemurs (Lemur catta) vary across natural and captive populations and correlate with environmental microbiota. Anim. Microbiome 2022, 4, 29. [Google Scholar] [CrossRef]
- McKenney, E.A.; O’Connell, T.M.; Rodrigo, A.; Yoder, A.D. Feeding strategy shapes gut metagenomic enrichment and functional specialization in captive lemurs. Gut Microbes 2018, 9, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Cuozzo, F.P.; Sauther, M.L.; Gould, L.; Sussman, R.W.; Villers, L.M.; Lent, C. Variation in dental wear and tooth loss among known-aged, older ring-tailed lemurs (Lemur catta): A comparison between wild and captive individuals. Am. J. Primatol. 2010, 72, 1026–1037. [Google Scholar] [CrossRef]
- Cuozzo, F.P.; Sauther, M.L. Severe wear and tooth loss in wild ring-tailed lemurs (Lemur catta): A function of feeding ecology, dental structure, and individual life history. J. Hum. Evol. 2006, 51, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Millette, J.B.; Sauther, M.L.; Cuozzo, F.P. Behavioral responses to tooth loss in wild ring-tailed lemurs (Lemur catta) at the Beza Mahafaly Special Reserve, Madagascar. Am. J. Phys. Anthr. 2009, 140, 120–134. [Google Scholar] [CrossRef]
- Collins, C.; Corkery, I.; Haigh, A.; McKeown, S.; Quirke, T.; O’Riordan, R. The effects of environmental and visitor variables on the behavior of free-ranging ring-tailed lemurs (Lemur catta) in captivity. Zoo. Biol. 2017, 36, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.D. Lemurs, in Exotic Animal Laboratory Diagnosis; Wiley: Hoboken, NJ, USA, 2020; pp. 229–240. [Google Scholar]
- Junge, R.E.; Williams, C.V.; Campbell, J. Nutrition and behavior of lemurs. Vet. Clin. N. Am. Exot. Anim. Pract. 2009, 12, 339–348, x-i. [Google Scholar] [CrossRef]
- Hansell, M.; Åsberg, A.; Laska, M. Food preferences and nutrient composition in zoo-housed ring-tailed lemurs, Lemur catta. Physiol. Behav. 2020, 226, 113125. [Google Scholar] [CrossRef]
- Worsley, S.F.; Videvall, E.; Harrison, X.A.; Björk, J.R.; Mazel, F.; Wanelik, K.M. Probing the functional significance of wild animal microbiomes using omics data. Funct. Ecol. 2024, 38, 2329–2349. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Hatfull, G.F.; Ford, M.E.; Smith, M.C.; Burns, R.N. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef]
- Chevallereau, A.; Pons, B.J.; van Houte, S.; Westra, E.R. Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed]
- Duller, S.; Moissl-Eichinger, C. Archaea in the Human Microbiome and Potential Effects on Human Infectious Disease. Emerg. Infect. Dis. 2024, 30, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Brugère, J.-F.; Gribaldo, S.; Schmitz, R.A.; Moissl-Eichinger, C. The host-associated archaeome. Nat. Rev. Microbiol. 2020, 18, 622–636. [Google Scholar] [CrossRef]
- Mohammadzadeh, R.; Mahnert, A.; Duller, S.; Moissl-Eichinger, C. Archaeal key-residents within the human microbiome: Characteristics, interactions and involvement in health and disease. Curr. Opin. Microbiol. 2022, 67, 102146. [Google Scholar] [CrossRef]
- Ufnar, J.A.; Wang, S.Y.; Ufnar, D.F.; Ellender, R.D. Methanobrevibacter ruminantium as an indicator of domesticated-ruminant fecal pollution in surface waters. Appl. Env. Microbiol. 2007, 73, 7118–7121. [Google Scholar] [CrossRef]
- Wei, Y.-Q.; Yang, H.-J.; Long, R.-J.; Wang, Z.-Y.; Cao, B.-B.; Ren, Q.-C.; Wu, T.-T. Characterization of natural co-cultures of Piromyces with Methanobrevibacter ruminantium from yaks grazing on the Qinghai-Tibetan Plateau: A microbial consortium with high potential in plant biomass degradation. AMB Express 2017, 7, 160. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, H.; Wang, Z.; Zhao, J.; Qi, H.; Wang, C.; Zhang, J.; Yang, T. Roughage biodegradation by natural co-cultures of rumen fungi and methanogens from Qinghai yaks. AMB Express 2022, 12, 123. [Google Scholar] [CrossRef]
- Grine, G.; Lotte, R.; Chirio, D.; Chevalier, A.; Raoult, D.; Drancourt, M.; Ruimy, R. Co-culture of Methanobrevibacter smithii with enterobacteria during urinary infection. EBioMedicine 2019, 43, 333–337. [Google Scholar] [CrossRef]
- Drancourt, M.; Djemai, K.; Gouriet, F.; Grine, G.; Loukil, A.; Bedotto, M.; Levasseur, A.; Lepidi, H.; Bou-Khalil, J.; Khelaifia, S.; et al. Methanobrevibacter smithii Archaemia in Febrile Patients with Bacteremia, Including Those with Endocarditis. Clin. Infect. Dis. 2021, 73, e2571–e2579. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Köhler, J.R.; Hube, B.; Puccia, R.; Casadevall, A.; Perfect, J.R. Fungi that Infect Humans. Microbiol. Spectr. 2017, 5, 813–843. [Google Scholar] [CrossRef] [PubMed]
- Strickland, A.B.; Shi, M. Mechanisms of fungal dissemination. Cell Mol. Life Sci. 2021, 78, 3219–3238. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-R.; Lin, C.-S.; Chang, C.-J.; Lin, T.-L.; Martel, J.; Ko, Y.-F.; Ojcius, D.M.; Lu, C.-C.; Young, J.D.; Lai, H.-C. Gut commensal Parabacteroides goldsteinii plays a predominant role in the anti-obesity effects of polysaccharides isolated from Hirsutella sinensis. Gut 2019, 68, 248–262. [Google Scholar] [CrossRef]
- Kim, N.Y.; Kim, S.; Park, H.M.; Lim, C.M.; Kim, J.; Park, J.Y.; Jeon, K.-B.; Poudel, A.; Lee, H.P.; Oh, S.-R.; et al. Cinnamomum verum extract inhibits NOX2/ROS and PKCδ/JNK/AP-1/NF-κB pathway-mediated inflammatory response in PMA-stimulated THP-1 monocytes. Phytomedicine 2023, 112, 154685. [Google Scholar] [CrossRef]
- Ansari, M.A.; Murali, M.; Prasad, D.; Alzohairy, M.A.; Almatroudi, A.; Alomary, M.N.; Udayashankar, A.C.; Singh, S.B.; Asiri, S.M.M.; Ashwini, B.S.; et al. Cinnamomum verum Bark Extract Mediated Green Synthesis of ZnO Nanoparticles and Their Antibacterial Potentiality. Biomolecules 2020, 10, 336. [Google Scholar] [CrossRef]
- Do, D.T.B.; Bui, T.H.; Phan, D.T.A. Persea Americana Mill seed extracts: Understanding insights into the antioxidant and antityrosinase activities and effects on preserving qualities of whiteleg shrimp (Litopenaus vannamei) during refrigerated storage. Food Chem. 2022, 373 Pt B, 131469. [Google Scholar] [CrossRef]
- Alajmi, F.; Al-Otaibi, T.; Al-Quraishy, S.; Al-Shaebi, E.M.; Al-Hoshani, N.; Dkhil, M.A.; Abdel-Gaber, R. Persea americana extract protects intestinal tissue from Eimeria papillata-induced murine Infection. BMC Vet. Res. 2023, 19, 248. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Prince, A.L.; Bader, D.; Hu, M.; Ganu, R.; Baquero, K.; Blundell, P.; Harris, R.A.; Frias, A.E.; Grove, K.L.; et al. High-fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model. Nat. Commun. 2014, 5, 3889. [Google Scholar] [CrossRef]
- Clayton, J.B.; Vangay, P.; Huang, H.; Ward, T.; Hillmann, B.M.; Al-Ghalith, G.A.; Travis, D.A.; Long, H.T.; Van Tuan, B.; Van Minh, V.; et al. Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. USA 2016, 113, 10376–10381. [Google Scholar] [CrossRef]
- Houtz, J.L.; Sanders, J.G.; Denice, A.; Moeller, A.H. Predictable and host-species specific humanization of the gut microbiota in captive primates. Mol. Ecol. 2021, 30, 3677–3687. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, X.; Zhang, M.; Pan, H. Comparative Analysis of Gut Microbiota between Wild and Captive Golden Snub-Nosed Monkeys. Animals 2023, 13, 1625. [Google Scholar] [CrossRef]
- Ning, Y.; Qi, J.; Dobbins, M.T.; Liang, X.; Wang, J.; Chen, S.; Ma, J.; Jiang, G. Comparative Analysis of Microbial Community Structure and Function in the Gut of Wild and Captive Amur Tiger. Front. Microbiol. 2020, 11, 1665. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; D’aigle, J.; Atadja, L.; Quaicoe, V.; Honarpisheh, P.; Ganesh, B.P.; Hassan, A.; Graf, J.; Petrosino, J.F.; Putluri, N.; et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Poststroke Recovery in Aged Mice. Circ. Res. 2020, 127, 453–465. [Google Scholar] [CrossRef]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.H. Butyrate producers, “The Sentinel of Gut”: Their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front. Microbiol. 2022, 13, 1103836. [Google Scholar] [CrossRef]
- Kang, X.; Liu, C.; Ding, Y.; Ni, Y.; Ji, F.; Lau, H.C.H.; Jiang, L.; Sung, J.J.Y.; Wong, S.H.; Yu, J. Roseburia intestinalis generated butyrate boosts anti-PD-1 efficacy in colorectal cancer by activating cytotoxic CD8(+) T cells. Gut 2023, 72, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Salanitro, J.P.; Muirhead, P.A.; Goodman, J.R. Morphological and physiological characteristics of Gemmiger formicilis isolated from chicken ceca. Appl. Env. Microbiol. 1976, 32, 623–632. [Google Scholar] [CrossRef]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; De Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015, 22, 971–982. [Google Scholar] [CrossRef]
- Wu, M.; Kasper, D.L. Fiber Sets up the Battleground for Intestinal Prevotella. Cell Host Microbe 2020, 28, 776–777. [Google Scholar] [CrossRef]
- Aravindraja, C.; Jeepipalli, S.; Vekariya, K.M.; Botello-Escalante, R.; Chan, E.K.; Kesavalu, L. Oral Spirochete Treponema denticola Intraoral Infection Reveals Unique miR-133a, miR-486, miR-126-3p, miR-126-5p miRNA Expression Kinetics during Periodontitis. Int. J. Mol. Sci. 2023, 24, 12105. [Google Scholar] [CrossRef]
- Ridgway, R.; Lu, H.; Blower, T.R.; Evans, N.J.; Ainsworth, S. Genomic and taxonomic evaluation of 38 Treponema prophage sequences. BMC Genom. 2024, 25, 549. [Google Scholar] [CrossRef] [PubMed]
- Dallas, J.W.; Warne, R.W. Captivity and Animal Microbiomes: Potential Roles of Microbiota for Influencing Animal Conservation. Microb. Ecol. 2023, 85, 820–838. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Wilmes, P. Human Gut Microbiome: Function Matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef]
- Karasov, W.H.; Martínez del Rio, C.; Caviedes-Vidal, E. Ecological physiology of diet and digestive systems. Annu. Rev. Physiol. 2011, 73, 69–93. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.Y.; Weitz, J.S. Not by (Good) Microbes Alone: Towards Immunocommensal Therapies. Trends Microbiol. 2019, 27, 294–302. [Google Scholar] [CrossRef]
- Rottenberg, H. The accelerated evolution of human cytochrome c oxidase—Selection for reduced rate and proton pumping efficiency? Biochim. Biophys. Acta Bioenerg. 2022, 1863, 148595. [Google Scholar] [CrossRef] [PubMed]
- Wardman, J.F.; Bains, R.K.; Rahfeld, P.; Withers, S.G. Carbohydrate-active enzymes (CAZymes) in the gut microbiome. Nat. Rev. Microbiol. 2022, 20, 542–556. [Google Scholar] [CrossRef]
- Amato, K.R.; Leigh, S.R.; Kent, A.; Mackie, R.I.; Yeoman, C.J.; Stumpf, R.M.; Wilson, B.A.; Nelson, K.E.; White, B.A.; Garber, P.A. The role of gut microbes in satisfying the nutritional demands of adult and juvenile wild, black howler monkeys (Alouatta pigra). Am. J. Phys. Anthr. 2014, 155, 652–664. [Google Scholar] [CrossRef]
- Amato, K.R.; Sanders, J.G.; Song, S.J.; Nute, M.; Metcalf, J.L.; Thompson, L.R.; Morton, J.T.; Amir, A.; McKenzie, V.J.; Humphrey, G.; et al. Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 2019, 13, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. J. Basic. Microbiol. 2021, 61, 1049–1070. [Google Scholar] [CrossRef]
- Nnadozie, C.F.; Odume, O.N. Freshwater environments as reservoirs of antibiotic resistant bacteria and their role in the dissemination of antibiotic resistance genes. Env. Pollut. 2019, 254 Pt B, 113067. [Google Scholar] [CrossRef]
- Shi, H.; Hu, X.; Li, W.; Zhang, J.; Hu, B.; Lou, L. Soil Component: A Potential Factor Affecting the Occurrence and Spread of Antibiotic Resistance Genes. Antibiotics 2023, 12, 333. [Google Scholar] [CrossRef]
- Zhang, A.-N.; Gaston, J.M.; Dai, C.L.; Zhao, S.; Poyet, M.; Groussin, M.; Yin, X.; Li, L.-G.; van Loosdrecht, M.C.M.; Topp, E.; et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 2021, 12, 4765. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Guo, X.; Li, L. Metagenomic Comparison of Gut Microbes of Lemur catta in Captive and Semi-Free-Range Environments. Animals 2025, 15, 1442. https://doi.org/10.3390/ani15101442
Xu C, Guo X, Li L. Metagenomic Comparison of Gut Microbes of Lemur catta in Captive and Semi-Free-Range Environments. Animals. 2025; 15(10):1442. https://doi.org/10.3390/ani15101442
Chicago/Turabian StyleXu, Chunzhong, Xinzi Guo, and Lian Li. 2025. "Metagenomic Comparison of Gut Microbes of Lemur catta in Captive and Semi-Free-Range Environments" Animals 15, no. 10: 1442. https://doi.org/10.3390/ani15101442
APA StyleXu, C., Guo, X., & Li, L. (2025). Metagenomic Comparison of Gut Microbes of Lemur catta in Captive and Semi-Free-Range Environments. Animals, 15(10), 1442. https://doi.org/10.3390/ani15101442