

Temporal Changes in Jejunal and Ileal Microbiota of Broiler Chickens with Clinical Coccidiosis (Eimeria maxima)

 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Husbandry and Tissue Sampling
2.2. Identification of Stem Cells and Morphological Measurements
2.3. DNA Isolation and 16s rRNA Library Preparation
2.4. Bioinformatics and Data Processing
2.5. Statistical Analysis
3. Results
3.1. Parameters Associated with Infection and Growth
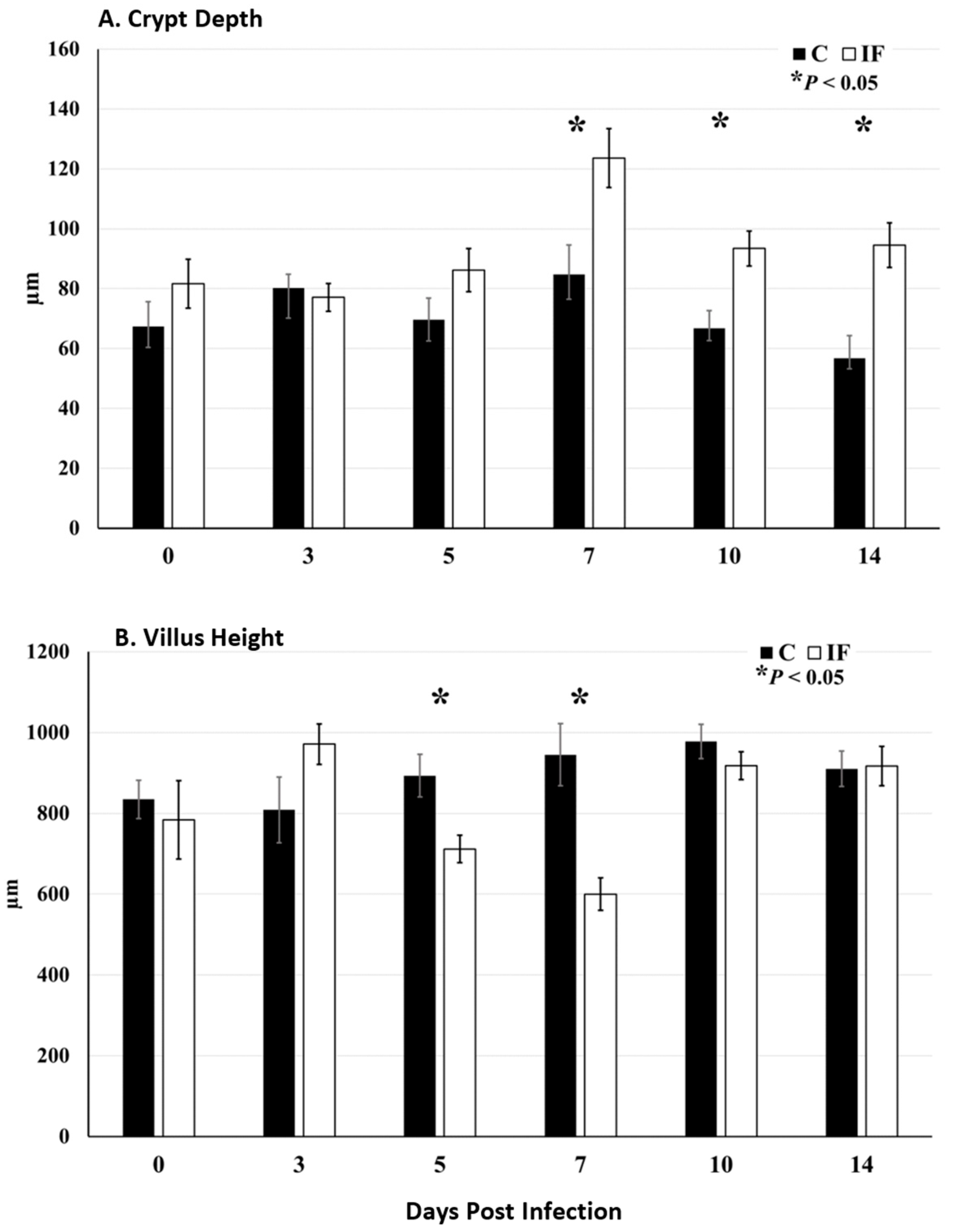
3.2. Gut Morphology
3.3. Sequencing Summary
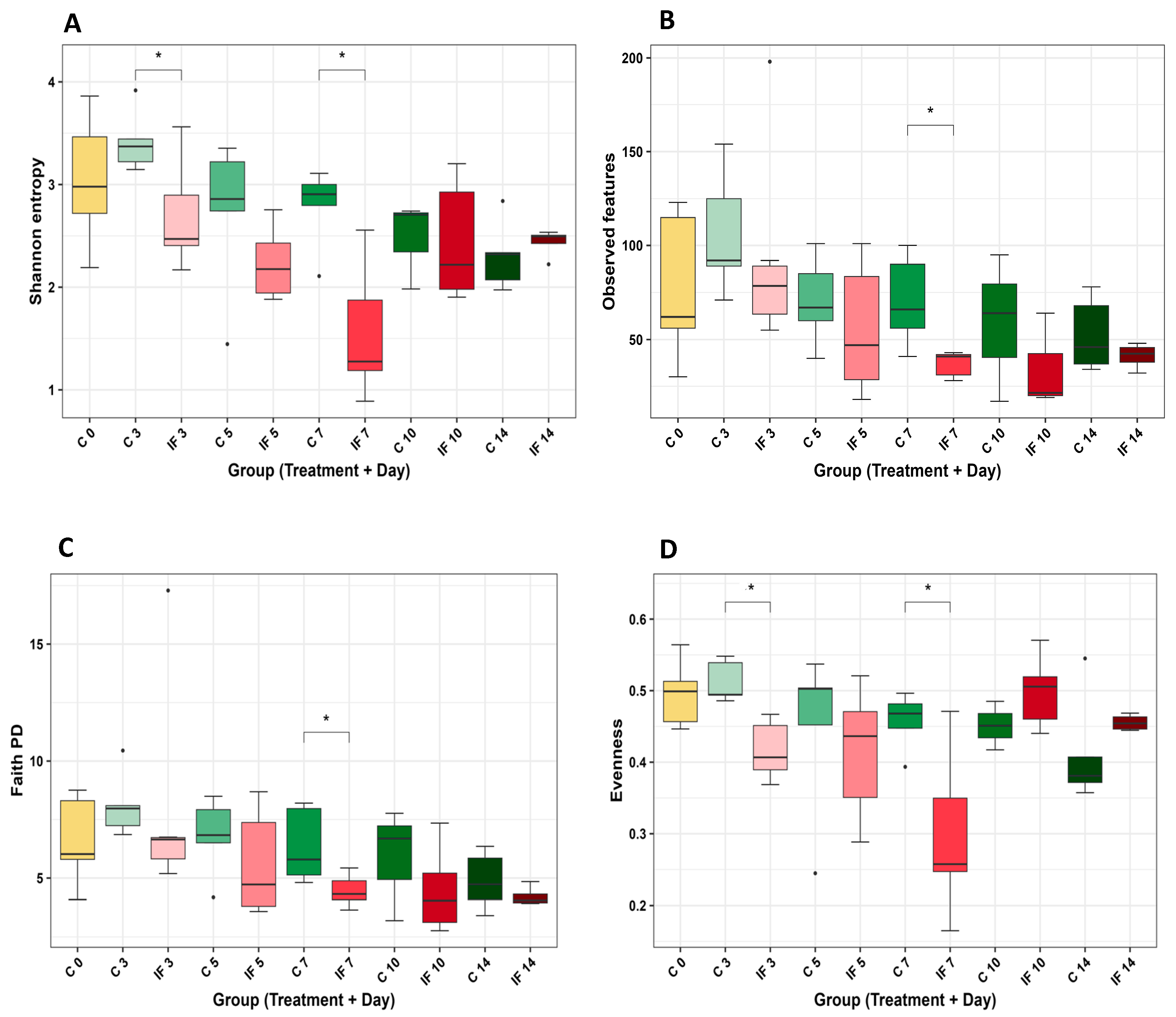
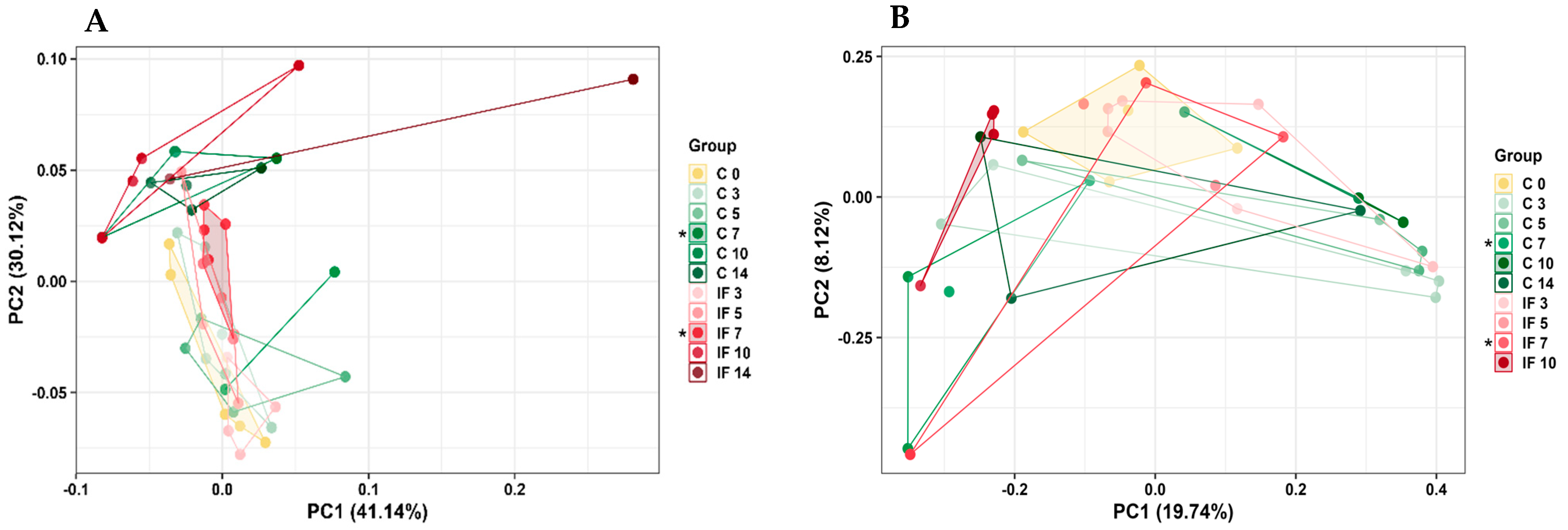
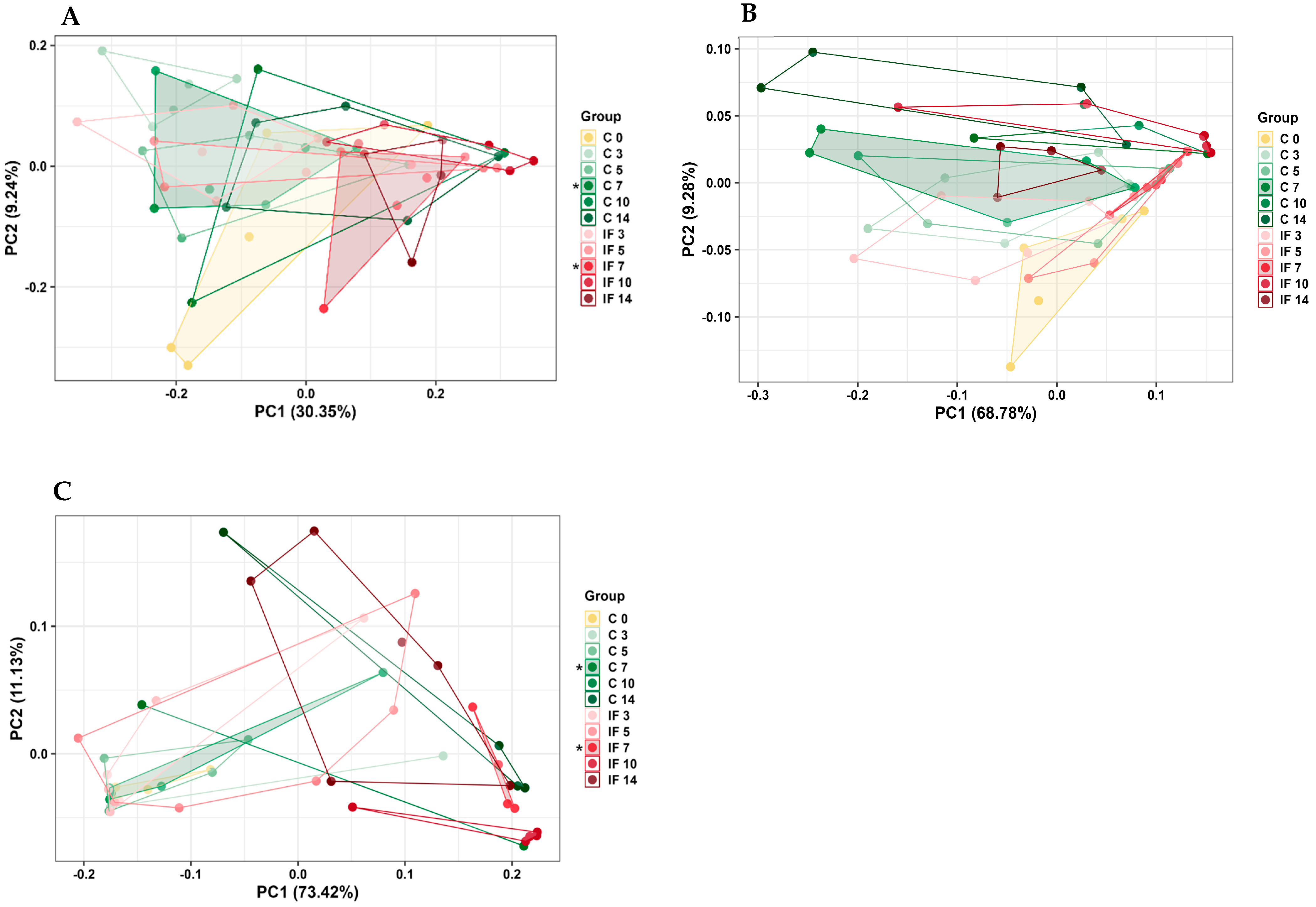
3.4. Alpha and Beta Diversity of the Microbiota of the Small Intestine
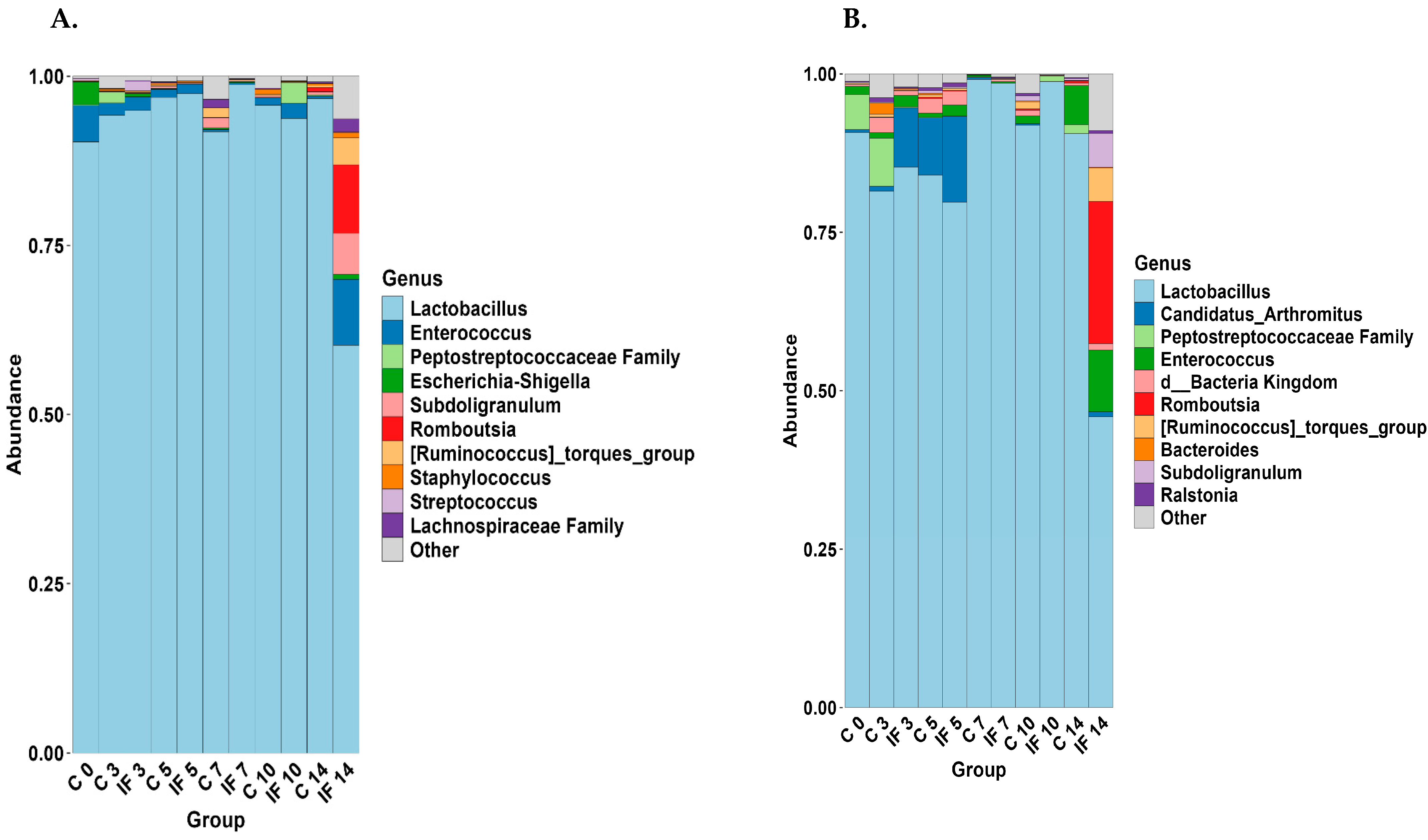
3.5. Taxonomy
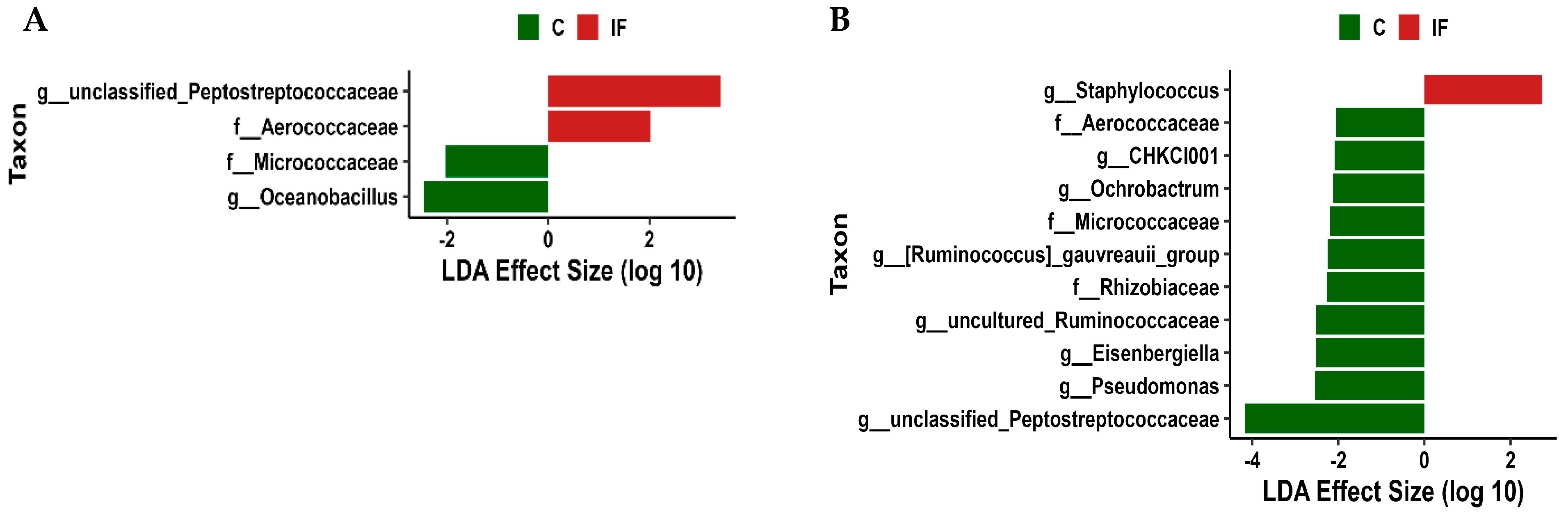
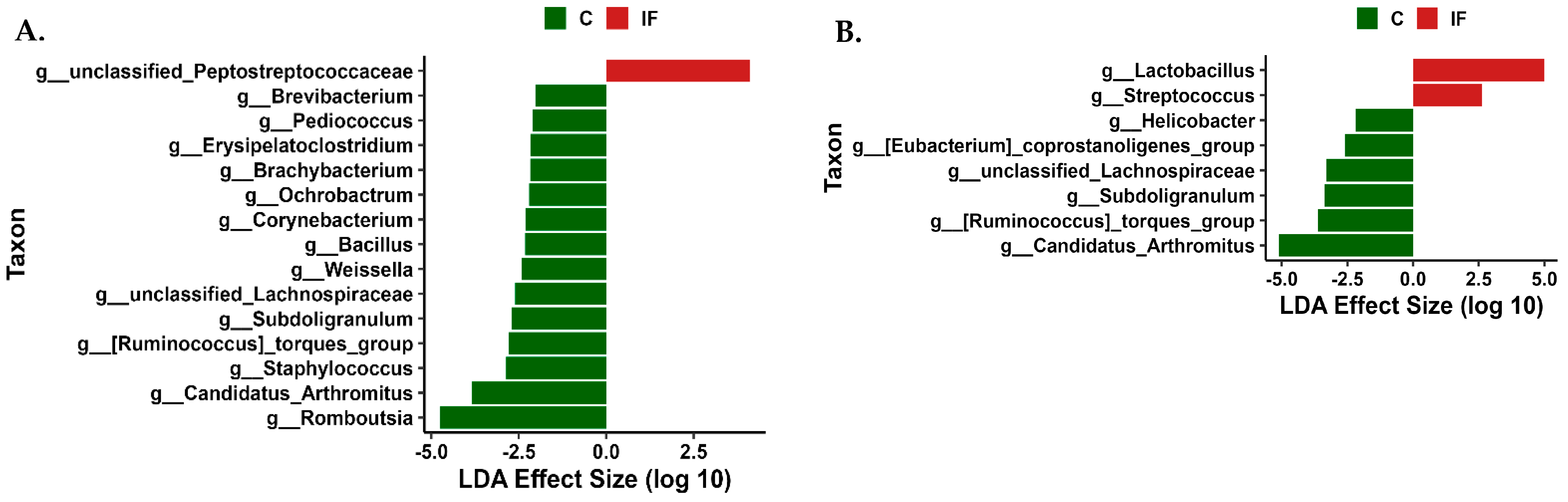
3.6. Differentially Abundant Taxa
3.7. Analysis of Predicted Functional Processes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mesa-Pineda, C.; Navarro-Ruíz, J.L.; López-Osorio, S.; Chaparro-Gutiérrez, J.J.; Gómez-Osorio, L.M. Chicken coccidiosis: From the parasite lifecycle to control of the disease. Front. Vet. Sci. 2021, 8, 787653. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.P.; Vrba, V.; Xia, D.; Jatau, I.D.; Spiro, S.; Nolan, M.J.; Underwood, G.; Tomley, F.M. Genetic and biological characterisation of three cryptic Eimeria operational taxonomic units that infect chickens (Gallus gallus domesticus). Int. J. Parasitol. 2021, 51, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Yu, Y.H.; Hua, K.F.; Chen, W.J.; Zaborski, D.; Dybus, A.; Hsiao, F.S.; Cheng, Y.H. Management and control of coccidiosis in poultry—A review. Anim. Biosci. 2024, 37, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Noack, S.; Chapman, H.D.; Selzer, P.M. Anticoccidial drugs of the livestock industry. Parasitol. Res. 2019, 118, 2009–2026. [Google Scholar] [CrossRef]
- Martins, R.R.; Silva, L.J.G.; Pereira, A.M.P.T.; Esteves, A.; Duarte, S.C.; Pena, A. Coccidiostats and poultry: A comprehensive review and current legislation. Foods 2022, 11, 2738. [Google Scholar] [CrossRef]
- Cloft, S.E.; Miska, K.B.; Jenkins, M.; Proszkowiec-Weglarz, M.; Kahl, S.; Wong, E.A. Temporal changes of genes associated with intestinal homeostasis in broiler chickens following a single infection with Eimeria acervulina. Poult. Sci. 2023, 102, 102537. [Google Scholar] [CrossRef]
- Elsasser, T.H.; Miska, K.; Kahl, S.; Fetterer, R.H.; Martínez Ramirez, A. Temporal pattern changes in duodenal protein tyrosine nitration events in response to Eimeria acervulina infection in chickens. J. Anim. Sci. 2018, 96, 2125–2138. [Google Scholar] [CrossRef]
- Rothwell, L.; Gramzinski, R.A.; Rose, M.E.; Kaiser, P. Avian coccidiosis: Changes in intestinal lymphocyte populations associated with the development of immunity to Eimeria maxima. Parasite Immunol. 1995, 17, 525–533. [Google Scholar] [CrossRef]
- Campos, P.M.; Miska, K.B.; Jenkins, M.C.; Yan, X.; Proszkowiec-Weglarz, M. Effects of Eimeria acervulina infection on the luminal and mucosal microbiota of the duodenum and jejunum in broiler chickens. Front. Microbiol. 2023, 14, 1147579. [Google Scholar] [CrossRef]
- Campos, P.M.; Miska, K.B.; Jenkins, M.C.; Yan, X.; Proszkowiec-Weglarz, M. Effects of Eimeria acervulina infection on the luminal and mucosal microbiota of the cecum and ileum in broiler chickens. Sci. Rep. 2024, 14, 10702. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Campos, P.M.; Darwish, N.; Shao, J.; Proszkowiec-Weglarz, M. Research Note: Choice of microbiota database affects data analysis and interpretation in chicken cecal microbiota. Poult. Sci. 2022, 101, 101971. [Google Scholar] [CrossRef] [PubMed]
- Borda-Molina, D.; Seifert, J.; Camarinha-Silva, A. Current Perspectives of the Chicken Gastrointestinal Tract and Its Microbiome. Comput. Struct. Biotechnol. J. 2018, 16, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.C. Physiological responses of chicken gut tissue to coccidial infection: Comparative effects of Eimeria acervulina and Eimeria mitis on mucosal mass, carotenoid content, and brush border enzyme activity. Poult. Sci. 1987, 66, 1306–1315. [Google Scholar] [CrossRef]
- Darwish, N.; Shao, J.; Schreier, L.L.; Proszkowiec-Weglarz, M. Choice of 16S ribosomal RNA primers affects the microbiome analysis in chicken ceca. Sci. Rep. 2021, 11, 11848. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—How do these taxonomies compare? BMC Genom. 2017, 18 (Suppl. 2), 114. [Google Scholar] [CrossRef]
- Robeson, M.S., 2nd; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing, Version 4.3.2; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 4 March 2024).
- Bisanz, J. qiime2R: Importing QIIME2 Artifacts and Associated Data into R Sessions, R Package Version 0.99; R Foundation for Statistical Computing: Vienna, Austria, 2018; Volume 13. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2020, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K.; Vaughan, D. dplyr: A Grammar of Data Manipulation, R Package Version 1.1.0; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://dplyr.tidyverse.org (accessed on 4 March 2024).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 4 March 2024).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Barnett, D.J.M.; Arts, I.C.W.; Penders, J. microViz: An R package for microbiome data visualization and statistics. J. Open Source Softw. 2021, 6, 3201. [Google Scholar] [CrossRef]
- Hansen, V.L.; Kahl, S.; Proszkowiec-Weglarz, M.; Jiménez, S.C.; Vaessen, S.F.C.; Schreier, L.L.; Jenkins, M.C.; Russell, B.; Miska, K.B. The effects of tributyrin supplementation on weight gain and intestinal gene expression in broiler chickens during Eimeria maxima-induced coccidiosis. Poult. Sci. 2021, 100, 100984. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.C.; Dubey, J.P.; Miska, K.; Fetterer, R. Differences in fecundity of Eimeria maxima strains exhibiting different levels of pathogenicity in its avian host. Vet. Parasitol. 2017, 236, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Conway, D.P.; Sasai, K.; Gaafar, S.M.; Smothers, C.D. Effects of different levels of oocyst inocula of Eimeria acervulina, E. tenella, and E. maxima on plasma constituents, packed cell volume, lesion scores, and performance in chickens. Avian Dis. 1993, 37, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Fetterer, R.H.; Barfield, R.C.; Jenkins, M.C. Protection of broiler chicks housed with immunized cohorts against infection with Eimeria maxima and E. acervulina. Avian Dis. 2015, 59, 98–105. [Google Scholar] [CrossRef]
- Sakkas, P.; Oikeh, I.; Blake, D.P.; Nolan, M.J.; Bailey, R.A.; Oxley, A.; Rychlik, I.; Lietz, G.; Kyriazakis, I. Does selection for growth rate in broilers affect their resistance and tolerance to Eimeria maxima? Vet. Parasitol. 2018, 258, 88–98. [Google Scholar] [CrossRef]
- Fernando, M.A.; McCraw, B.M. Mucosal morphology and cellular renewal in the intestine of chickens following a single infection of Eimeria acervulina. J. Parasitol. 1973, 59, 493–501. [Google Scholar] [CrossRef]
- Choi, J.; Tompkins, Y.H.; Teng, P.-Y.; Gogal, R.M., Jr.; Kim, W.K. Effects of Tannic Acid Supplementation on Growth Performance, Oocyst Shedding, and Gut Health of in Broilers Infected with Eimeria maxima. Animals 2022, 12, 1378. [Google Scholar] [CrossRef]
- Choi, J.; Kim, W. Interactions of Microbiota and Mucosal Immunity in the Ceca of Broiler Chickens Infected with Eimeria tenella. Vaccines 2022, 10, 1941. [Google Scholar] [CrossRef]
- Zhou, B.H.; Jia, L.S.; Wei, S.S.; Ding, H.Y.; Yang, J.Y.; Wang, H.W. Effects of Eimeria tenella infection on the barrier damage and microbiota diversity of chicken cecum. Poult. Sci. 2020, 99, 1297–1305. [Google Scholar] [CrossRef]
- Cha, J.O.; Zhao, J.; Yang, M.S.; Kim, W.I.; Cho, H.S.; Lim, C.W.; Kim, B. Oocyst-Shedding Patterns of Three Eimeria Species in Chickens and Shedding Pattern Variation Depending on the Storage Period of Eimeria tenella Oocysts. J. Parasitol. 2018, 104, 18–22. [Google Scholar] [CrossRef]
- Stanley, D.; Wu, S.B.; Rodgers, N.; Swick, R.A.; Moore, R.J. Differential responses of cecal microbiota to fishmeal, Eimeria and Clostridium perfringens in a necrotic enteritis challenge model in chickens. PLoS ONE 2014, 9, e104739. [Google Scholar] [CrossRef]
- Wu, S.B.; Stanley, D.; Rodgers, N.; Swick, R.A.; Moore, R.J. Two necrotic enteritis predisposing factors, dietary fishmeal and Eimeria infection, induce large changes in the caecal microbiota of broiler chickens. Vet. Microbiol. 2014, 169, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.; Yitbarek, A.; Snyder, R.; Patterson, R.; Barta, J.R.; Karrow, N.; Kiarie, E. Responses of broiler chickens to I challenge when fed a nucleotide-rich yeast extract. Poult. Sci. 2019, 98, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Proszkowiec-Weglarz, M.; Miska, K.B.; Schreier, L.L.; Grim, C.J.; Jarvis, K.G.; Shao, J.; Vaessen, S.; Sygall, R.; Jenkins, M.C.; Kahl, S.; et al. Research Note: Effect of butyric acid glycerol esters on ileal and cecal mucosal and luminal microbiota in chickens challenged with Eimeria maxima. Poult. Sci. 2020, 99, 5143–5148. [Google Scholar] [CrossRef] [PubMed]
- Marková, K.; Kreisinger, J.; Vinkler, M. Are there consistent effects of gut microbiota composition on performance, productivity and condition in poultry? Poult. Sci. 2024, 103, 103752. [Google Scholar] [CrossRef]
- Latorre, J.D.; Adhikari, B.; Park, S.H.; Teague, K.D.; Graham, L.E.; Mahaffey, B.D.; Baxter, M.F.A.; Hernandez-Velasco, X.; Kwon, Y.M.; Ricke, S.C.; et al. Evaluation of the epithelial barrier function and ileal microbiome in an established necrotic enteritis challenge model in broiler chickens. Front. Vet. Sci. 2018, 5, 199. [Google Scholar] [CrossRef]
- Miska, K.B.; Proszkowiec-Weglarz, M.; Buiatte, V.; Mahmoud, M.; Lesko, T.; Jenkins, M.C.; Chopra, S.; Lorenzoni, G. Comparative analysis of microbiota in the ceca of broiler chickens with necrotic enteritis fed a commercial corn diet or with corn high in flavonoids (PennHFD1). Front. Microbiomes 2023, 2, 1212130. [Google Scholar] [CrossRef]
- Tierney, J.; Gowing, H.; Van Sinderen, D.; Flynn, S.; Stanley, L.; McHardy, N.; Hallahan, S.; Mulcahy, G. In vitro inhibition of Eimeria tenella invasion by indigenous chicken Lactobacillus species. Vet. Parasitol. 2004, 122, 171–182. [Google Scholar] [CrossRef]
- Mirsalami, S.M.; Mirsalami, M. Effects of duo-strain probiotics on growth, digestion, and gut health in broiler chickens. Vet. Anim. Sci. 2023, 24, 100343. [Google Scholar] [CrossRef]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef]
- Onrust, L.; Ducatelle, R.; Van Driessche, K.; De Maesschalck, C.; Vermeulen, K.; Haesebrouck, F.; Eeckhaut, V.; Van Immerseel, F. Steering Endogenous Butyrate Production in the Intestinal Tract of Broilers as a Tool to Improve Gut Health. Front. Vet. Sci. 2015, 2, 75. [Google Scholar] [CrossRef] [PubMed]
- Basbas, C.; Aly, S.; Okello, E.; Karle, B.M.; Lehenbauer, T.; Williams, D.; Ganda, E.; Wiedmann, M.; Pereira, R.V. Effect of Intramammary Dry Cow Antimicrobial Treatment on Fresh Cow’s Milk Microbiota in California Commercial Dairies. Antibiotics 2022, 11, 963. [Google Scholar] [CrossRef] [PubMed]
- Igartua, C.; Davenport, E.R.; Gilad, Y.; Nicolae, D.L.; Pinto, J.; Ober, C. Host genetic variation in mucosal immunity pathways influences the upper airway microbiome. Microbiome 2017, 5, 16. [Google Scholar] [CrossRef]
- Feng, X.; Li, T.; Zhu, H.; Liu, L.; Bi, S.; Chen, X.; Zhang, H. Effects of challenge with Clostridium perfringens, Eimeria and both on ileal microbiota of yellow feather broilers. Front. Microbiol. 2022, 13, 1063578. [Google Scholar] [CrossRef]
- Lages da Silva, D.H.; Marques da Silva, R.L.; Rios, D.L.; de Souza, D.D.G.; Aburjaile, F.; de Freitas Neto, O.C.; Camargos Lara, L.J.; Dias Araújo, M.; Ecco, R. Intestinal microbiota diversity from broilers with runting and stunting syndrome performed by metagenomics. Avian Pathol. 2024, 53, 408–418. [Google Scholar] [CrossRef]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 2020, 8, 46. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| J-C | J-M | IL-C | IL-M | |
|---|---|---|---|---|
| Number of samples | 60 | 60 | 60 | 60 |
| Raw reads | 6,321,969 | 1,906,686 | 8,520,563 | 3,669,527 |
| DADA2 --p-trunc-len-f | 269 | 235 | 269 | 271 |
| DADA2 --p-trunc-len-r | 217 | 205 | 229 | 208 |
| Reads after DADA2 | 5,212,955 | 1,312,059 | 6,443,444 | 2,663,546 |
| Reads after filtering | 5,060,831 | 1,158,586 | 6,434,195 | 2,652,074 |
| Reads per sample (range) | 114–256,509 | 81–255,261 | 13,847–257,570 | 197–503,642 |
| Mean reads per sample | 84,347 | 19,310 | 107,237 | 44,201 |
| Total number of ASVs | 1001 | 1273 | 984 | 1371 |
| ASV read length (range) | 269–465 | 234–427 | 269–473 | 271–466 |
| Mean ASV read length | 418 | 358 | 420 | 385 |
| Sequencing depth | 27,462 | 5658 | 49,158 | 13,067 |
| Day Post-Infection Infection Status | BW (kg) | BWG 2 (kg) | FI (kg) | FCR | |
|---|---|---|---|---|---|
| 3 PI | C | 1.12 | 0.23 f | 1.36 | 1.49 c |
| IF | 1.17 | 0.22 f | 1.37 | 1.55 c | |
| 5 PI | C | 1.23 | 0.38 e | 2.40 | 1.58 c |
| IF | 1.24 | 0.37 e | 2.30 | 1.57 c | |
| 7 PI | C | 1.33 | 0.61 d | 3.72 | 1.54 c |
| IF | 1.50 | 0.45 e | 3.77 | 2.13 ab | |
| 10 PI | C | 1.51 | 0.78 c | 5.77 | 1.85 abc |
| IF | 2.05 | 0.63 d | 5.51 | 2.27 a | |
| 14 PI | C | 1.95 | 1.19 a | 7.85 | 1.65 bc |
| IF | 2.05 | 1.03 b | 7.27 | 1.79 abc | |
| SEM 3 | 0.12 | 0.03 | 0.29 | 0.11 | |
| Main effect day PI | |||||
| 3 PI | 1.15 b | 0.23 e | 1.36 e | 1.52 b | |
| 5 PI | 1.23 b | 0.37 d | 2.35 d | 1.57 b | |
| 7 PI | 1.41 b | 0.53 c | 3.75 c | 1.83 ab | |
| 10 PI | 1.78 a | 0.70 b | 5.64 b | 2.06 a | |
| 14 PI | 2.00 a | 1.11 a | 7.56 a | 1.72 b | |
| SEM 3 | 0.08 | 0.02 | 0.20 | 0.08 | |
| Main effect infection status | |||||
| C | 1.43 b | 0.64 a | 4.22 | 1.62 b | |
| IF | 1.60 a | 0.54 b | 4.04 | 1.86 a | |
| SEM 3 | 0.05 | 0.01 | 0.13 | 0.05 | |
| Analysis of variance | Probabilities 4 | ||||
| Day post-infection × infection status | 0.18 | 0.0053 | 0.82 | 0.0487 | |
| Day post-infection | <0.0001 | <0.0001 | <0.0001 | 0.0001 | |
| Infection status | 0.0246 | <0.0001 | 0.33 | 0.0015 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miska, K.B.; Campos, P.M.; Cloft, S.E.; Jenkins, M.C.; Proszkowiec-Weglarz, M. Temporal Changes in Jejunal and Ileal Microbiota of Broiler Chickens with Clinical Coccidiosis (Eimeria maxima). Animals 2024, 14, 2976. https://doi.org/10.3390/ani14202976
Miska KB, Campos PM, Cloft SE, Jenkins MC, Proszkowiec-Weglarz M. Temporal Changes in Jejunal and Ileal Microbiota of Broiler Chickens with Clinical Coccidiosis (Eimeria maxima). Animals. 2024; 14(20):2976. https://doi.org/10.3390/ani14202976
Chicago/Turabian StyleMiska, Katarzyna B., Philip M. Campos, Sara E. Cloft, Mark C. Jenkins, and Monika Proszkowiec-Weglarz. 2024. "Temporal Changes in Jejunal and Ileal Microbiota of Broiler Chickens with Clinical Coccidiosis (Eimeria maxima)" Animals 14, no. 20: 2976. https://doi.org/10.3390/ani14202976
APA StyleMiska, K. B., Campos, P. M., Cloft, S. E., Jenkins, M. C., & Proszkowiec-Weglarz, M. (2024). Temporal Changes in Jejunal and Ileal Microbiota of Broiler Chickens with Clinical Coccidiosis (Eimeria maxima). Animals, 14(20), 2976. https://doi.org/10.3390/ani14202976