
Does the Colonizing Population Exhibit a Reduced Genetic Diversity and Allele Surfing? A Case Study of the Midday Gerbil (Meriones meridianus Pallas) Expanding Its Range

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
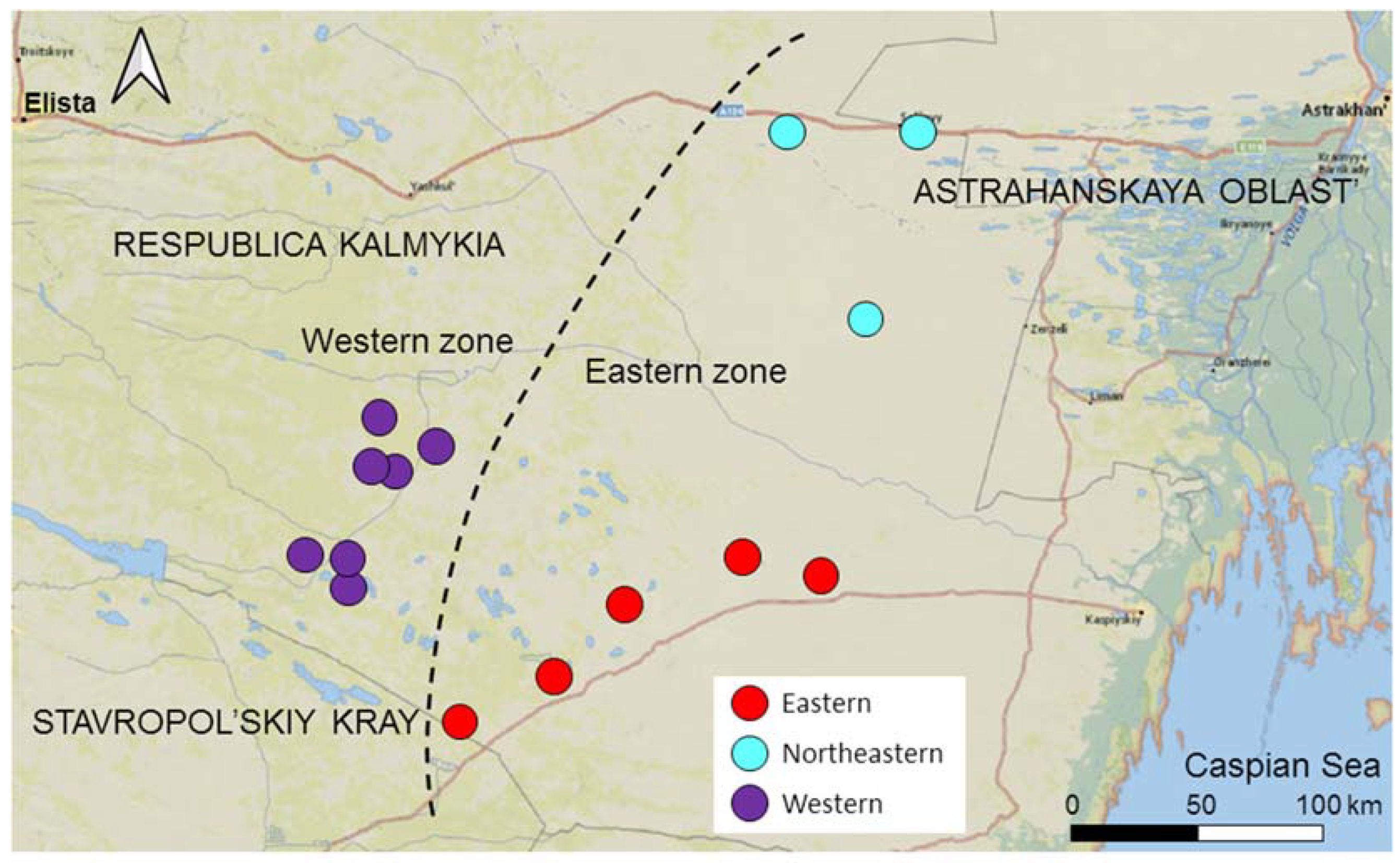
2.1. Study Area
2.2. Data Collection
2.3. Ethics Statement
2.4. DNA Extraction and Amplification
2.5. Data Analysis
3. Results
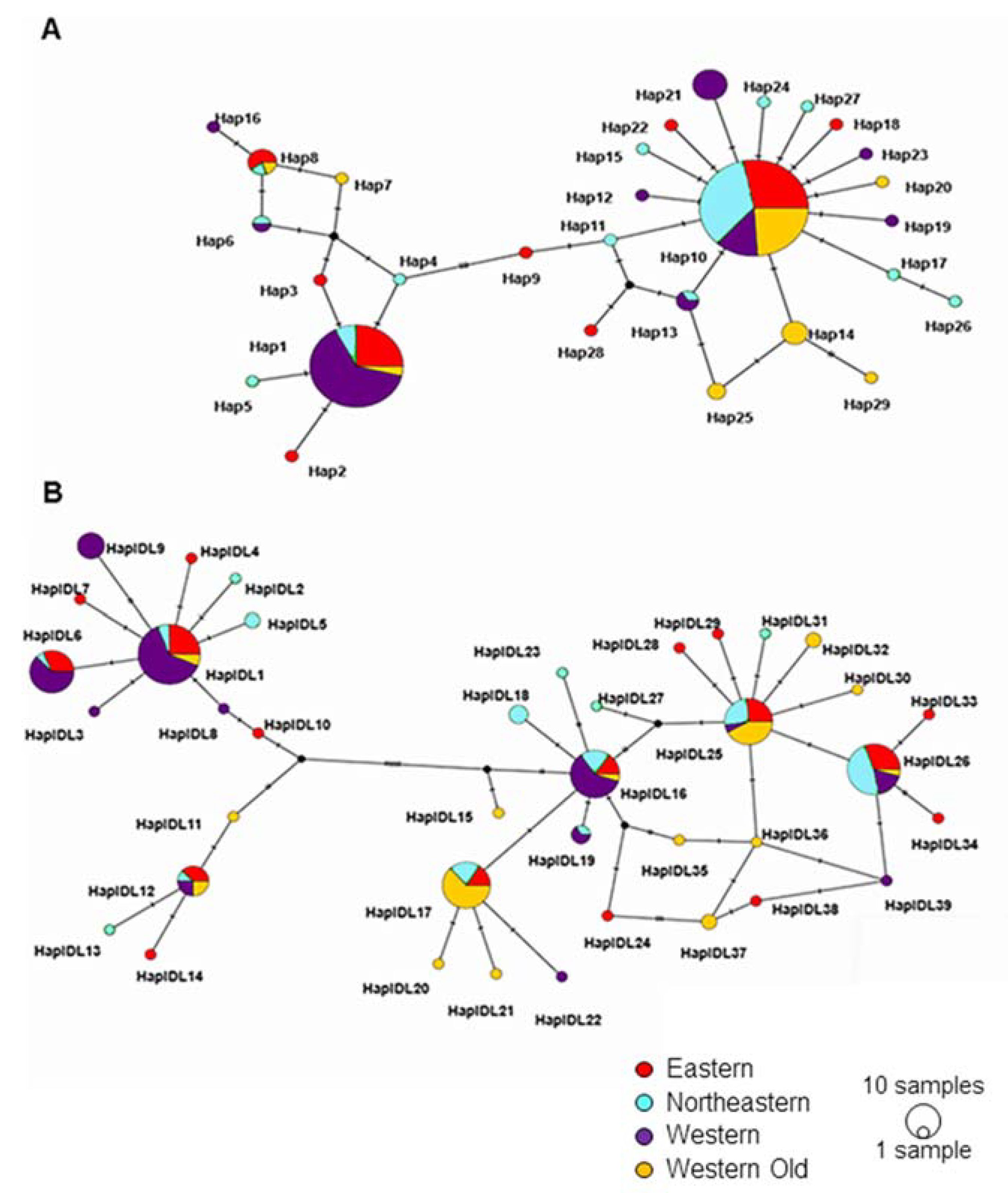
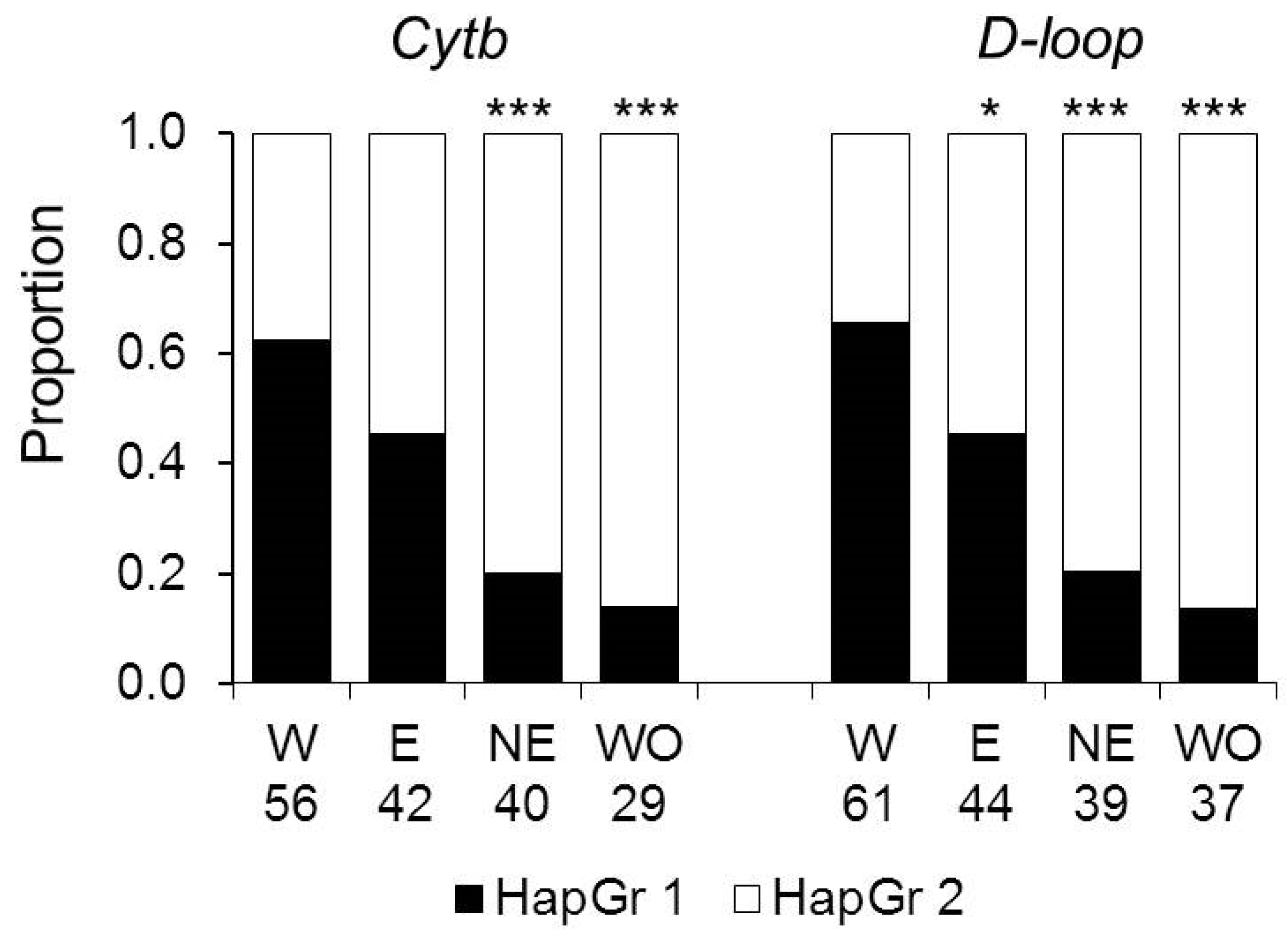
3.1. Genetic Diversity in the Current Population
3.2. Genetic Diversity and Differentiation between Subpopulations
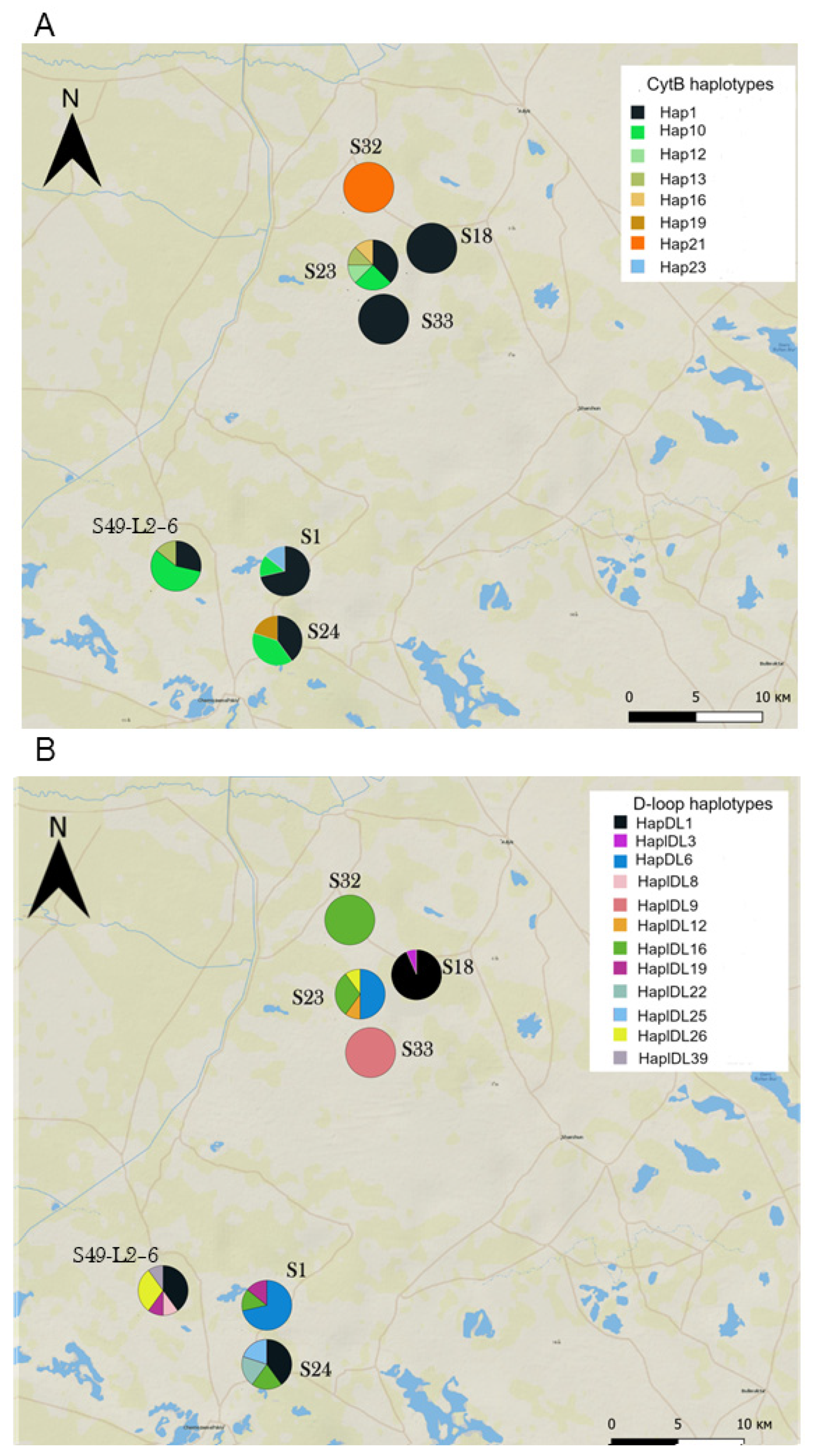
3.3. Genetic Diversity and Differentiation within the Colonizing Subpopulation
4. Discussion
4.1. The Midday Gerbil Population Shows Signs of Demographic and Spatial Expansion
4.2. Genetic Specificity of the Recently Established Colonist Subpopulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sakai, A.K.; Allendorf, F.W.; Holt, J.S.; Lodge, D.M.; Molofsky, J.; With, K.A.; Baughman, S.; Cabin, R.J.; Cohen, J.E.; Ellstrand, N.C.; et al. The population biology of invasive species. Annu. Rev. Ecol. Syst. 2021, 32, 305–332. [Google Scholar] [CrossRef]
- Welles, S.R.; Dlugosch, K.M. Population genomics of colonization and invasion. In Population Genomics: Concepts, Approaches and Applications; Rajora, O.P., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 655–683. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Schierenbeck, K.A. Hybridization as a stimulus for the evolution of invasiveness in plants? Proc. Natl. Acad. Sci. USA 2020, 97, 7043–7050. [Google Scholar] [CrossRef] [PubMed]
- Facon, B.; Hufbauer, R.A.; Tayeh, A.; Loiseau, A.; Lombaert, E.; Vitalis, R.; Guillemaud, T.; Lundgren, J.G.; Estoup, A. Inbreeding depression is purged in the invasive insect Harmonia axyridis. Curr. Biol. 2011, 21, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Ochocki, B.M.; Miller, T.E.X. Rapid evolution of dispersal ability makes biological invasions faster and more variable. Nat. Commun. 2017, 8, 14315. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.K.; Ochocki, B.M.; Crawford, K.M.; Compagnoni, A.; Miller, T.E.X. Genetic mixture of multiple source populations accelerates invasive range expansion. J. Anim. Ecol. 2017, 86, 21–34. [Google Scholar] [CrossRef]
- Williams, J.L.; Hufbauer, R.A.; Miller, T.E.X. How evolution modifies the variability of range expansion. Trends Ecol. Evol. 2019, 34, 903–913. [Google Scholar] [CrossRef]
- Excoffier, L.; Ray, N. Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol. Evol. 2008, 23, 347–351. [Google Scholar] [CrossRef]
- Excoffier, L.; Foll, M.; Petit, R.J. Genetic consequences of range expansions. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 481–501. [Google Scholar] [CrossRef]
- Graciá, E.; Botella, F.; Anadón, J.D.; Edelaar, P.; Harris, D.J.; Giménez, A. Surfing in tortoises? Empirical signs of genetic structuring owing to range expansion. Biol. Lett. 2013, 9, 20121091. [Google Scholar] [CrossRef]
- White, T.A.; Perkins, S.E.; Heckel, G.; Searle, J.B. Adaptive evolution during an ongoing range expansion: The invasive bank vole (Myodes glareolus) in Ireland. Mol. Ecol. 2013, 22, 2971–2985. [Google Scholar] [CrossRef]
- Chuang, A.; Peterson, C.R. Expanding population edges: Theories, traits, and trade-offs. Glob. Chang. Biol. 2016, 22, 494–512. [Google Scholar] [CrossRef] [PubMed]
- Rougemont, Q.; Leroy, T.; Rondeau, E.B.; Koop, B.; Bernatchez, L. Allele surfing causes maladaptation in a Pacific salmon of conservation concern. PLoS Genet. 2023, 19, e1010918. [Google Scholar] [CrossRef] [PubMed]
- Schrey, A.W.; Liebl, A.L.; Richards, C.L.; Martin, L.B. Range expansion of house sparrows (Passer domesticus) in Kenya: Evidence of genetic admixture and human-mediated dispersal. J. Hered. 2014, 105, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Bubac, C.M.; Spellman, G.M. How connectivity shapes genetic structure during range expansion: Insights from the Virginia’s Warbler. Auk 2016, 133, 213–230. [Google Scholar] [CrossRef]
- Birzu, G.; Matin, S.; Hallatschek, O.; Korolev, K.S. Genetic drift in range expansions is very sensitive to density dependence in dispersal and growth. Ecol. Lett. 2019, 22, 1817–1827. [Google Scholar] [CrossRef]
- Uller, T.; Leimu, R. Founder events predict changes in genetic diversity during human-mediated range expansions. Glob. Chang. Biol. 2011, 17, 3478–3485. [Google Scholar] [CrossRef]
- Schmidt, C.; Dray, S.; Garroway, C.J. Genetic and species-level biodiversity patterns are linked by demography and ecological opportunity. Evolution 2022, 76, 86–100. [Google Scholar] [CrossRef]
- Ibrahim, K.M.; Nichols, R.A.; Hewitt, G.M. Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 1996, 77, 282–291. [Google Scholar] [CrossRef]
- Neronov, V.V.; Tchabovskii, A.V.; Aleksandrov, D.Y.; Kasatkin, M.V. Spatial distribution of rodents under conditions of anthropogenic dynamics of vegetation in the South of Kalmykia. Russ. J. Ecol. 1997, 28, 328–334. [Google Scholar]
- Shenbrot, G.I.; Krasnov, B.R.; Rogovin, K.A. Spatial Ecology of Desert Rodent Communities; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Nanova, O.G.; Lebedev, V.S.; Matrosova, V.A.; Adiya, Y.; Undrakhbayar, E.; Surov, A.V.; Shenbrot, G.I. Phylogeography, phylogeny, and taxonomical revision of the Midday jird (Meriones meridianus) species complex from Dzungaria. J. Zool. Syst. Evol. Res. 2020, 58, 1335–1358. [Google Scholar] [CrossRef]
- Varshavsky, S.N.; Popov, N.V.; Varshavsky, B.S.; Shilov, M.N.; Tikhomirov, E.L.; Bugakov, A.A. Modification of the species composition of rodents in the north-western region of the Caspian Sea area under the effect of anthropogenic factors. Zool. Zhurnal 1991, 70, 92–100. (In Russian) [Google Scholar]
- Shilova, S.A.; Chabovskiĭ, A.V.; Isaev, S.I.; Neronov, V.V. Dynamics of rodent community and populations in Kalmyk semideserts under decreasing pasture load and increasing humidity. Izv. Akad. Nauk. Ser. Biol. 2000, 3, 332–344. (In Russian) [Google Scholar]
- Tchabovsky, A.V.; Savinetskaya, L.E.; Surkova, E.N.; Ovchinnikova, N.L.; Kshnyasev, I.A. Delayed threshold response of a rodent population to human-induced landscape change. Oecologia 2016, 182, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Tchabovsky, A.; Savinetskaya, L.; Surkova, E. Breeding versus survival: Proximate causes of abrupt population decline under environmental change in a desert rodent, the midday gerbil (Meriones meridianus). Integr. Zool. 2019, 14, 366–375. [Google Scholar] [CrossRef]
- Surkova, E.N.; Kulik, A.A.; Kuznetsova, E.V.; Bazykina, S.G.; Savinetskaya, L.E.; Tchabovsky, A.V. Chyornye Zemli of Kalmykia: Is the desert returning back? Priroda 2022, 8, 13–20. (In Russian) [Google Scholar]
- Tchabovsky, A.V.; Surkova, E.N.; Savinetskaya, L.E.; Kulik, A.A. Range expansion and population patterns on the wave of colonization: The Midday gerbil (Meriones meridianus Pallas 1773, Muridae, Rodentia) in Kalmykia taken as a model. Biol. Bull. Russ. Acad. Sci. 2023, 50, 2552–2560. [Google Scholar] [CrossRef]
- Schipanov, N.A. Universal live trap for small mammals. Zool. Zhurnal 1987, 66, 759–761. (In Russian) [Google Scholar]
- Surkova, E.N.; Savinetskaya, L.E.; Khropov, I.S.; Tchabovsky, A.V. Flexible males, reactive females: Faecal glucocorticoid metabolites indicate increased stress in the colonist population, damping with time in males but not in females. J. Comp. Physiol. B 2024, 194, 545–554. [Google Scholar] [CrossRef]
- ASAB/ABS Guidelines for the ethical treatment of nonhuman animals in behavioural research and teaching. Anim. Behav. 2024, 207, I–XI. [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Pres: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Wang, Y.; Zhao, L.M.; Fang, F.J.; Liao, J.C.; Liu, N.F. Intraspecific molecular phylogeny and phylogeography of the Meriones meridianus (Rodentia: Cricetidae) complex in northern China reflect the processes of desertification and the Tianshan Mountains uplift. Biol. J. Linn. Soc. Lond. 2013, 110, 362–383. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Soriano, A.; Ramos-Onsins, S.E.; Rozas, J.; Calafell, F.; Navarro, A. Statistical Power analysis of neutrality tests under demographic expansions, contractions and bottlenecks with recombination. Genetics 2008, 179, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics, 1st ed.; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 1992, 132, 619–633. [Google Scholar] [CrossRef]
- Clement, M.; Snell, Q.; Walke, P.; Posada, D.; Crandall, K. TCS: Estimating gene genealogies. Parallel Distrib. Process. Symp. Int. 2002, 2, 184. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Ricotta, C. Can we trust the chord (and the Hellinger) distance? Community Ecol. 2019, 20, 104–106. [Google Scholar] [CrossRef]
- Zou, Y.; Axmacher, J.C. The chord-normalized expected species shared (CNESS)-distance represents a superior measure of species turnover patterns. Methods Ecol. Evol. 2020, 11, 273–280. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electronica 2001, 4, 4. [Google Scholar]
- Habib, K.A.; Jeong, D.; Myoung, J.G.; Kim, M.S.; Jang, Y.S.; Shim, J.S.; Lee, Y.H. Population genetic structure and demographic history of the fat greenling Hexagrammos otakii. Genes. Genomics 2011, 33, 413–423. [Google Scholar] [CrossRef]
- Gerlach, G.; Jueterbock, A.; Kraemer, P.; Deppermann, J.; Harmand, P. Calculations of population differentiation based on GST and D: Forget GST but not all of statistics! Mol. Ecol. 2010, 19, 3845–3852. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. R Package Version 2.6-8. 2024. Available online: https://CRAN.R-project.org/package=vegan (accessed on 19 September 2024).
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, R.A.; Badyaev, A.V. Coupling of dispersal and aggression facilitates the rapid range expansion of a passerine bird. Proc. Natl. Acad. Sci. USA 2007, 104, 15017–15022. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, R.A.; Kruuk, L.E.B. Evolution of genetic integration between dispersal and colonization ability in a bird. Evolution 2009, 63, 968–977. [Google Scholar] [CrossRef]
- Clobert, J.; Le Galliard, J.F.; Cote, J.; Meylan, S.; Massot, M. Informed dispersal, heterogeneity in animal dispersal syndromes and the dynamics of spatially structured populations. Ecol. Lett. 2009, 12, 197–209. [Google Scholar] [CrossRef]
- Eccard, J.A.; Mazza, V.; Holland, C.; Stuart, P. The timid invasion: Behavioural adjustments and range expansion in a non-native rodent. Proc. R. Soc. Lond. B 2023, 290, 20230823. [Google Scholar] [CrossRef]
- Edmonds, C.A.; Lillie, A.S.; Cavalli-Sforza, L.L. Mutations arising in the wave front of an expanding population. Proc. Natl. Acad. Sci. USA 2004, 101, 975–979. [Google Scholar] [CrossRef]
- Klopfstein, S.; Currat, M.; Excoffier, L. The fate of mutations surfing on the wave of a range expansion. Mol. Biol. Evol. 2006, 23, 482–490. [Google Scholar] [CrossRef]
- Nichols, R.A.; Hewitt, G.M. The genetic consequences of long distance dispersal during colonization. Heredity 1994, 72, 312–317. [Google Scholar] [CrossRef]
- Shilova, S.A.; Tchabovsky, A.V. Population response of rodents to control with rodenticides. Curr. Zool. 2009, 55, 81–91. [Google Scholar] [CrossRef]
- Guisan, A.; Thuiller, W. Predicting species distribution: Offering more than simple habitat models. Ecol. Lett. 2005, 8, 993–1009. [Google Scholar] [CrossRef] [PubMed]
- Antonovics, J.; McKane, A.J.; Newman, T.J. Spatiotemporal dynamics in marginal populations. Am. Nat. 2006, 167, 16–27. [Google Scholar] [CrossRef]
- Eckert, C.G.; Samis, K.E.; Lougheed, S.C. Genetic variation across species’ geographical ranges: The central-marginal hypothesis and beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef]
- Millette, K.L.; Keyghobadi, N. The relative influence of habitat amount and configuration on genetic structure across multiple spatial scales. Ecol. Evol. 2015, 5, 73–86. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | N | h | S | Eta | Hd | Pi | k | V | Fu’s Fs | Tajima’s D |
|---|---|---|---|---|---|---|---|---|---|---|
| Cytb | 138 | 24 | 23 | 23 | 0.716 | 0.0031 | 3.554 | 0.0007 | −6.45 * | −0.426 p > 0.10 |
| D-loop | 144 | 30 | 32 | 32 | 0.896 | 0.0080 | 7.468 | 0.0001 | −2.92 * | −0.164 p > 0.10 |
| Subpopulation | N | h | S | Hd | Pi | hpr | Npr/N | Fu’s Fs | Tajima’s D |
|---|---|---|---|---|---|---|---|---|---|
| Cytb | |||||||||
| Eastern | 42 | 9 | 12 | 0.685 | 0.003 | 6 | 0.14 | 0.703 | 0.879 |
| Northeastern | 40 | 13 | 15 | 0.608 | 0.002 | 8 | 0.20 | −4.077 *** | −0.905 |
| Western | 56 | 9 | 14 | 0.619 | 0.003 | 5 | 0.20 | 1.078 | 0.349 |
| Western Old | 29 | 8 | 13 | 0.645 | 0.002 | 5 | 0.31 | −0.739 | −0.915 |
| D-loop | |||||||||
| Eastern | 44 | 17 | 23 | 0.918 | 0.008 | 10 | 0.23 | −0.903 | 1.420 |
| Northeastern | 39 | 15 | 25 | 0.889 | 0.007 | 7 | 0.26 | −1.132 * | 0.139 |
| Western | 61 | 12 | 23 | 0.823 | 0.007 | 5 | 0.16 | 2.383 ** | 0.983 |
| Western Old | 37 | 15 | 22 | 0.846 | 0.005 | 9 | 0.30 | −2.260 *** | −0.148 |
| Subpopulation 1 | Subpopulation 2 | K1 | K2 | K1,2 | FST | Dxy | Da | K2P |
|---|---|---|---|---|---|---|---|---|
| Cytb Current subpopulations | ||||||||
| Northeastern | Western | 2.524 | 3.405 | 4.034 | 0.265 | 0.0035 | 0.00094 | 0.0035 |
| Eastern | Western | 3.592 | 3.405 | 3.688 | 0.051 | 0.0032 | 0.00017 | 0.0032 |
| Northeastern | Eastern | 2.524 | 3.592 | 3.321 | 0.079 | 0.0029 | 0.00023 | 0.0029 |
| Cytb Current subpopulations versus extinct subpopulation | ||||||||
| Eastern | Western Old | 3.592 | 2.399 | 3.489 | 0.141 | 0.0031 | 0.00043 | 0.0032 |
| Northeastern | Western Old | 2.524 | 2.399 | 2.482 | 0.008 | 0.0022 | 0.00002 | 0.0022 |
| Western | Western Old | 3.405 | 2.399 | 4.336 | 0.331 | 0.0038 | 0.00125 | 0.0038 |
| D-loop Current subpopulations | ||||||||
| Northeastern | Western | 6.157 | 6.449 | 8.616 | 0.268 | 0.0092 | 0.00247 | 0.0025 |
| Eastern | Western | 7.570 | 6.449 | 7.516 | 0.067 | 0.0080 | 0.00054 | 0.0005 |
| Northeastern | Eastern | 6.157 | 7.570 | 7.400 | 0.073 | 0.0079 | 0.00057 | 0.0006 |
| D-loop Current subpopulations versus extinct subpopulation | ||||||||
| Eastern | Western Old | 7.570 | 5.081 | 7.345 | 0.139 | 0.0078 | 0.00109 | 0.0011 |
| Northeastern | Western Old | 6.157 | 5.081 | 5.686 | 0.012 | 0.0061 | 0.00007 | 0.0001 |
| Western | Western Old | 6.449 | 5.081 | 8.701 | 0.337 | 0.0093 | 0.00313 | 0.0032 |
| Population | Distance, km (min–max) | FST (min–max) | Variance FST | P | Chord (min–max) | Variance Chord |
|---|---|---|---|---|---|---|
| Cytb | ||||||
| Western | 18.6 (1.1–33.7) | 0.389 (0.000–1.000) | 0.11 | 0.48 | 0.86 (0.00–1.41) | 0.19 |
| Eastern | 73.0 (12.9–148.5) | 0.046 (0.000–0.359) | 0.01 | 0.07 | 0.60 (0.30–1.06) | 0.04 |
| Western Old | 2.7 (0.9–3.6) | 0.113 (0.000–0.232) | 0.01 | 0.67 | 0.91 (0.21–1.26) | 0.36 |
| D-loop | ||||||
| Western | 0.425 (0.000–1.000) | 0.11 | 0.81 | 1.22 (0.45–1.41) | 0.09 | |
| Eastern | 0.038 (0.000–0.282) | 0.01 | 0.14 | 1.06 (0.67–1.41) | 0.04 | |
| Western Old | 0.216 (0.099–0.398) | 0.03 | 0.67 | 1.13 (0.92–1.41) | 0.06 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batova, O.N.; Markov, N.I.; Titov, S.V.; Tchabovsky, A.V. Does the Colonizing Population Exhibit a Reduced Genetic Diversity and Allele Surfing? A Case Study of the Midday Gerbil (Meriones meridianus Pallas) Expanding Its Range. Animals 2024, 14, 2720. https://doi.org/10.3390/ani14182720
Batova ON, Markov NI, Titov SV, Tchabovsky AV. Does the Colonizing Population Exhibit a Reduced Genetic Diversity and Allele Surfing? A Case Study of the Midday Gerbil (Meriones meridianus Pallas) Expanding Its Range. Animals. 2024; 14(18):2720. https://doi.org/10.3390/ani14182720
Chicago/Turabian StyleBatova, Olga N., Nikolay I. Markov, Sergey V. Titov, and Andrey V. Tchabovsky. 2024. "Does the Colonizing Population Exhibit a Reduced Genetic Diversity and Allele Surfing? A Case Study of the Midday Gerbil (Meriones meridianus Pallas) Expanding Its Range" Animals 14, no. 18: 2720. https://doi.org/10.3390/ani14182720
APA StyleBatova, O. N., Markov, N. I., Titov, S. V., & Tchabovsky, A. V. (2024). Does the Colonizing Population Exhibit a Reduced Genetic Diversity and Allele Surfing? A Case Study of the Midday Gerbil (Meriones meridianus Pallas) Expanding Its Range. Animals, 14(18), 2720. https://doi.org/10.3390/ani14182720