
Gut Microbiota Profiling as a Promising Tool to Detect Equine Inflammatory Bowel Disease (IBD)
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Background Information on Horses with IBD
2.2. DNA Extraction and Sequencing
2.3. Sequence Processing
2.4. Calprotectin Extraction
2.5. Statistical Analyses of the Sequence Data
2.5.1. Alpha- and Beta-Diversity
2.5.2. Differentially Abundant OTUs
2.5.3. Artificial Neural Network Model
3. Results
3.1. Microbiota Composition
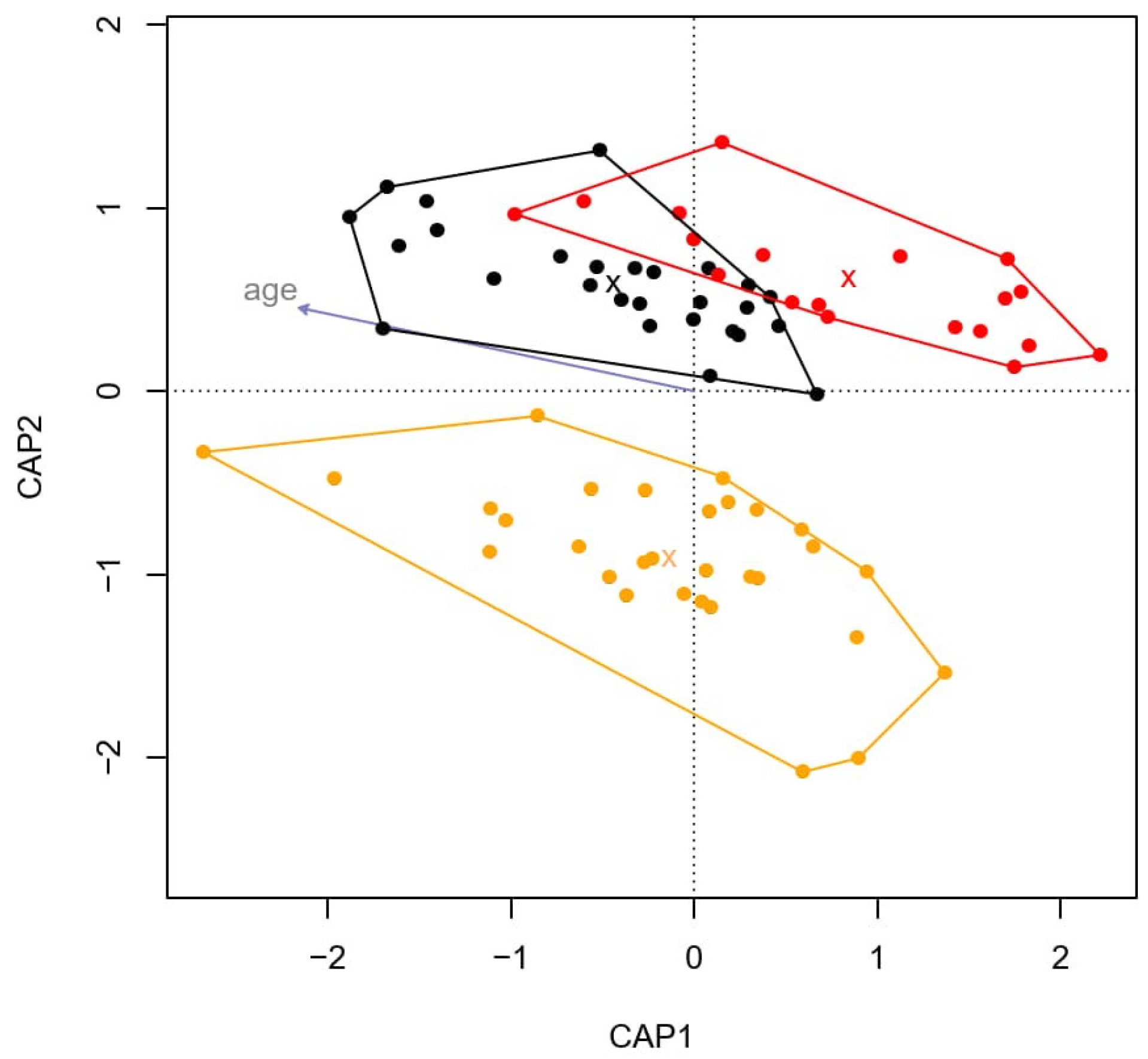
3.2. Differences in Microbiota Composition between Healthy Horses and Horses with IBD
3.3. Differentially Abundant OTUs
3.4. Prediction of IBD Prevalence with the Artificial Neural Network Model
3.5. Fecal Calprotectin Measurement
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuo, T.; Kamm, M.A.; Colombel, J.F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Berlow, M.; Phillips, J.N.; Derryberry, E.P. Effects of urbanization and landscape on gut microbiomes in white-crowned sparrows. Microb. Ecol. 2021, 81, 253–266. [Google Scholar] [CrossRef]
- Cui, G.; Liu, H.; Xu, G.; Laugsand, J.B.; Pang, Z. Exploring links between industrialization, urbanization, and Chinese inflammatory bowel disease. Front. Med. 2021, 8, 757025. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto-Hill, S.; Alenghat, T. Inflammation-associated microbiota composition across domestic animals. Front. Genet. 2021, 12, 649599. [Google Scholar] [CrossRef]
- Qin, X. Etiology of inflammatory bowel disease: A unified hypothesis. World J. Gastroenterol. 2012, 18, 1708. [Google Scholar] [CrossRef] [PubMed]
- Kofla-Dłubacz, A.; Pytrus, T.; Akutko, K.; Sputa-Grzegrzółka, P.; Piotrowska, A.; Dziegel, P. Etiology of IBD—is it still a mystery? Int. J. Mol. Sci. 2022, 23, 12445. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, J. Gut microbiota and inflammatory bowel disease. WIREs Mech. Dis. 2021, 14, e1540. [Google Scholar] [CrossRef]
- Zhang, Y.; Si, X.; Yang, L.; Wang, H.; Sun, Y.; Liu, N. Association between intestinal microbiota and inflammatory bowel disease. Anim. Models Exp. Med. 2022, 5, 311–322. [Google Scholar] [CrossRef]
- Doulidis, P.G.; Galler, A.I.; Hausmann, B.; Berry, D.; Rodríguez-Rojas, A.; Burgener, I.A. Gut microbiome signatures of Yorkshire Terrier enteropathy during disease and remission. Sci. Rep. 2023, 13, 4337. [Google Scholar] [CrossRef]
- Ning, L.; Zhou, Y.-L.; Sun, H.; Zhang, Y.; Shen, C.; Wang, Z.; Hong, J. Microbiome and metabolome features in inflammatory bowel disease via multi-omics integration analyses across cohorts. Nat. Commun. 2023, 14, 7135. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E. Role of gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Di Vicenzo, F.; Del Gaudio, A.; Petito, V.; Lopetuso, L.R.; Scaldaferri, F. Gut microbiota, intestinal permeability, and systemic inflammation: A narrative review. Intern. Emerg. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Marsilio, S.; Pilla, R.; Sarawichitr, B.; Chow, B.; Hill, S.L.; Ackermann, M.R.; Scot Estep, J.; Lidbury, J.A.; Steiner, J.M.; Suchodolski, J.S. Characterization of the fecal microbiome in cats with inflammatory bowel disease or alimentary small cell lymphoma. Sci. Rep. 2019, 9, 19208. [Google Scholar] [CrossRef]
- Vitale, V. Inflammatory bowel disease in horses: What do we know? Equine Vet. Educ. 2022, 34, 493–500. [Google Scholar] [CrossRef]
- Feary, D.J.; Hassel, D.M. Enteritis and colitis in horses. Vet. Clin. N. Am. Equine Pract. 2006, 22, 437–479. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, S.E.; Weese, J.S.; Adams, A.A. Comparison of the fecal microbiota in horses with equine metabolic syndrome and metabolically normal controls fed a similar all-forage diet. J. Equine Vet. Sci. 2016, 44, 9–16. [Google Scholar] [CrossRef]
- Arnold, C.E.; Pilla, R.; Chaffin, M.K.; Leatherwood, J.L.; Wickersham, T.A.; Callaway, T.R.; Lawhon, S.D.; Lidbury, J.A.; Steiner, J.M.; Suchodolski, J.S. The effects of signalment, diet, geographic location, season, and colitis associated with antimicrobial use or Salmonella infection on the fecal microbiome of horses. J. Vet. Intern. Med. 2021, 35, 2437–2448. [Google Scholar] [CrossRef]
- McKinney, C.A.; Bedenice, D.; Pacheco, A.P.; Oliveira, B.C.M.; Paradis, M.-R.; Mazan, M.; Widmer, G. Assessment of clinical and microbiota responses to fecal microbial transplantation in adult horses with diarrhea. PLoS ONE 2021, 16, e0244381. [Google Scholar] [CrossRef]
- Tedjo, D.I.; Smolinska, A.; Savelkoul, P.H.; Masclee, A.A.; van Schooten, F.J.; Pierik, M.J.; Jonkers, D.M.A.E. The fecal microbiota as a biomarker for disease activity in Crohn’s disease. Sci. Rep. 2016, 6, 35216. [Google Scholar] [CrossRef] [PubMed]
- Öhman, L.; Lasson, A.; Strömbeck, A.; Isaksson, S.; Hesselmar, M.; Simrén, M.; Strid, H.; Magnusson, M.K. Fecal microbiota dynamics during disease activity and remission in newly diagnosed and established ulcerative colitis. Sci. Rep. 2021, 11, 8641. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-Y.; Park, J.-L.; Yeo, M.-K.; Kang, S.-B.; Kim, J.-M.; Kim, J.S.; Kim, S.-Y. Diagnosis of Chron’s disease and ulcerative colitis using the microbiome. BMC Microbiol. 2023, 23, 336. [Google Scholar] [CrossRef] [PubMed]
- Kauter, A.; Epping, L.; Semmler, T.; Antao, E.-M.; Kannapin, D.; Stoeckle, S.D.; Gehlen, H.; Lübke-Becker, A.; Günther, S.; Wieler, L.H.; et al. The gut microbiome of horses: Current research on equine enteral microbiota and future perspectives. Anim. Microbiome 2019, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, F.S.; Burri, E.; Beglinger, C. The role and utility of faecal markers in inflammatory bowel disease. Therap. Adv. Gastroenterol. 2015, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Jukic, A.; Bakiri, L.; Wagner, E.F.; Tilg, H.; Adolph, T.E. Calprotectin: From biomarker to biological function. Gut 2021, 70, 1978–1988. [Google Scholar] [CrossRef]
- Khaki-Khatibi, F.; Qujeq, D.; Kashifard, M.; Moein, S.; Maniati, M.; Vaghari-Tabari, M. Calprotectin in inflammatory bowel disease. Clin. Chim. Acta. 2020, 510, 556–565. [Google Scholar] [CrossRef]
- Heilmann, R.M.; Berghoff, N.; Mansell, J.; Grützner, N.; Parnell, N.K.; Gurtner, C.; Steiner, J.M. Association of fecal calprotectin concentrations with disease severity, response to treatment, and other biomarkers in dogs with chronic inflammatory enteropathies. J. Vet. Intern. Med. 2018, 32, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Riggers, D.S.; Xenoulis, P.G.; Karra, D.A.; Enderle, L.L.; Köller, G.; Böttcher, D.; Heilmann, R.M. Fecal calprotectin concentrations in cats with chronic enteropathies. Vet. Sci. 2023, 10, 419. [Google Scholar] [CrossRef]
- Faleiros, R.R.; Nuovo, G.J.; Belknap, J.K. Calprotectin in myeloid and epithelial cells of laminae from horses with black walnut extract-induced laminitis. J. Vet. Intern. Med. 2009, 23, 174–181. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Zavia, L.S.; Gomez, D.E.; Caddey, B.B.; Boerlin, P.; Surette, M.G.; Arroyo, L.G. Direct and culture-enriched 16S rRNA sequencing of cecal content of healthy horses and horses with typhlocolitis. PLoS ONE 2023, 18, e0284193. [Google Scholar]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abenet, C.C.; Al-Ghalith, G.A.; Caporaso, J.G. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Kuhn, M.; Wing, J.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; Team, R.C.; et al. Package ‘caret’. R J. 2020, 223, 48. [Google Scholar]
- Ripley, B.; Venables, W.; Ripley, M.B. Package ‘nnet’. R Package Version 2016, 7, 3–12. [Google Scholar]
- Chaucheyras-Durand, F.; Sacy, A.; Karges, K.; Apper, E. Gastro-intestinal microbiota in equines and its role in health and disease: The black box opens. Microorganisms 2022, 10, 2517. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Huttenhower, C. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Pisani, A.; Rausch, P.; Bang, C.; Ellul, S.; Tabone, T.; Marantidis Cordina, C.; Zahra, G.; Franke, A.; Ellul, P. Dysbiosis in the gut microbiota in patients with inflammatory bowel disease during remission. Microbiol. Spectr. 2022, 10, e00611622. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Liu, H.; Yu, H.; Chen, M.; Yang, T.; Zeng, X.; Qiao, S. Core altered micro-organisms in colitis mouse model: A comprehensive time-point and fecal microbiota transplantation analysis. Antibiotics 2021, 10, 643. [Google Scholar] [CrossRef]
- Derakhshani, H.; De Buck, J.; Mortier, R.; Barkema, H.W.; Krause, D.O.; Khafipour, E. The features of fecal and ileal mucosa-associated microbiota in dairy calves during early infection with Mycobacterium avium subspecies paratuberculosis. Front. Microbiol. 2016, 7, 426. [Google Scholar] [CrossRef]
- Lee, S.M.; Park, H.T.; Park, S.; Lee, J.H.; Kim, D.; Yoo, H.S.; Kim, D. A machine learning approach reveals a microbiota signature for infection with Mycobacterium avium subsp. paratuberculosis in cattle. Microbiol. Spectr. 2023, 11, e03134-22. [Google Scholar] [CrossRef]
- Matthews, C.; Walsh, A.M.; Gordon, S.V.; Markey, B.; Cotter, P.D.; O’Mahony, J. Differences in faecal microbiome taxonomy, diversity and functional potential in a bovine cohort experimentally challenged with Mycobacterium avium subsp. paratuberculosis (MAP). Animals 2023, 13, 1652. [Google Scholar] [CrossRef] [PubMed]
- Amos, G.C.A.; Sergaki, C.; Logan, A.; Iriarte, R.; Bannaga, A.; Chandrapalan, S.; Arasaradnam, R.P. Exploring how microbiome signatures change across inflammatory bowel disease conditions and disease locations. Sci. Rep. 2021, 11, 18699. [Google Scholar] [CrossRef] [PubMed]
- Kedia, S.; Ghosh, T.S.; Jain, S.; Desigamani, A.; Kumar, A.; Gupta, V.; Bopanna, S.; Yadav, D.P.; Goyal, S.; Makharia, G.; et al. Gut microbiome diversity in acute severe colitis is distinct from mild to moderate ulcerative colitis. J. Gastroenterol. Hepatol. 2021, 36, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, L.I.H.; Morgan, X.C. Searching for a consensus among inflammatory bowel disease studies: A systematic meta-analysis. Inflamm. Bowel Dis. 2023, 29, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Chen, K.-Y.; Mueller, O.; Zhang, H.; Rakhilin, N.; Chen, J.; Shen, X. Microbiota of inflammatory bowel disease models. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2018, 2018, 2374–2377. [Google Scholar]
- Kozik, A.J.; Nakatsu, C.H.; Chun, H.; Jones-Hall, Y.L. Comparison of the fecal, cecal, and mucus microbiome in male and female mice after TNBS-induced colitis. PLoS ONE 2019, 14, e0225079. [Google Scholar] [CrossRef] [PubMed]
- Axelrad, J.E.; Cadwell, K.H.; Colombel, J.-F.; Shah, S.C. The role of gastrointestinal pathogens in inflammatory bowel disease: A systematic review. Ther. Adv. Gastroenterol. 2021, 14, 1–17. [Google Scholar] [CrossRef]
- Shan, Y.; Lee, M.; Chang, E.B. The gut microbiome and inflammatory bowel disease. Annu. Rev. Med. 2022, 27, 455–468. [Google Scholar] [CrossRef]
- El Hadad, J.; Schreiner, P.; Vavricka, S.R.; Greuter, T. The genetics of inflammatory bowel disease. Mol. Diagn. Ther. 2024, 28, 27–35. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Heilmann, R.M.; Paital, B.; Patel, A.; Yadav, V.K.; Wong, D.; Jergens, A.E. Oxidative stress, hormones, and effects of natural antioxidants on intestinal inflammation in inflammatory bowel disease. Front. Endocrinol. 2023, 14, 1217165. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, X.; Liu, C.; Su, L.; Xia, Z.; Li, X.; Xiao, L. Dysbiosis of the fecal microbiota in the TNBS-induced Chron’s disease mouse model. Appl. Microbiol. Biotechnol. 2016, 100, 4485–4494. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 103. [Google Scholar] [CrossRef]
- Dahal, R.M.; Kim, S.; Kim, Y.K.; Kim, E.S.; Kim, J. Insight into gut dysbiosis of patients with inflammatory bowel disease and ischemic colitis. Front. Microbiol. 2023, 14, 1174832. [Google Scholar] [CrossRef]
- Boshuizen, B.; Ploeg, M.; Dewulf, J.; Klooster, S.; de Bruijn, M.; Picavet, M.-T.; Palmers, K.; Plancke, L.; De Cock, H.; Theelen, M.; et al. Inflammatory bowel disease (IBD) in horses: A retrospective study exploring the value of different diagnostic approaches. BMC Vet. Res. 2018, 14, 21. [Google Scholar] [CrossRef]
- Hattori, S.; Nakamura, M.; Yamamura, T.; Maeda, K.; Sawada, T.; Mizutani, Y.; Fujishiro, M. The microbiome can predict mucosal healing in small intestine in patients with Chron’s disease. J. Gastroenterol. 2020, 55, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Olbjørn, C.; Cvancarova-Småstuen, M.; Fossum Moen, A.E. Targeted analysis of the gut microbiome for diagnosis, prognosis and treatment individualization in pediatric inflammatory bowel disease. Microorganisms 2022, 10, 1273. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Sharon, I.; Martín Quijada, N.; Pasolli, E.; Fabbrini, M.; Vitali, F.; Agamennone, V.; Turroni, S. The core human microbiome: Does it exist and how can we find it? A critical review of the concept. Nutrients 2022, 14, 2872. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Champomier Vergés, M.-C.; Charles, T.; Schloter, M. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar]
- Stewart, H.L.; Pitta, D.; Indugu, N.; Vecchiarelli, B.; Hennessy, M.L.; Engiles, J.B.; Southwood, L.L. Changes in the fecal bacterial microbiota during hospitalization of horses with colic and the effect of different causes of colic. Equine Vet. J. 2021, 53, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrette, P.; Balamurugan, R. The Firmicutes/Bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Cheong, H.; Yoon, J.; Kim, A.; Yun, Y.; Unno, T. Comparison of the fecal microbiota of horses with intestinal disease and their healthy counterparts. Vet. Sci. 2021, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Whitfield-Cargile, C.M.; Chamoun-Emanuelli, A.M.; Cohen, N.D.; Richardson, L.M.; Ajami, N.J.; Dockery, H.J. Differential effects of selective and non-selective cyclooxygenase inhibitors on fecal microbiota in adult horses. PLoS ONE 2018, 13, e0202527. [Google Scholar] [CrossRef] [PubMed]
- Kunz, I.G.; Reed, K.J.; Metcalf, J.L.; Hassel, D.M.; Coleman, R.J.; Hess, T.M.; Coleman, S.J. Equine fecal microbiota changes associated with anthelmintic administration. J. Equine Veter. Sci. 2019, 77, 98–106. [Google Scholar] [CrossRef]
- Weese, J.S.; Holcombe, S.J.; Embertson, R.M.; Kurtz, K.A.; Roessner, H.A.; Jalali, M.; Wismer, S.E. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Veter. J. 2015, 47, 641–649. [Google Scholar] [CrossRef]
- Arroyo, L.G.; Rossi, L.; Santos, B.P.; Gomez, D.E.; Surette, M.G.; Costa, M.C. Luminal and mucosal microbiota of the cecum and large colon of healthy and diarrheic horses. Animals 2020, 10, 1403. [Google Scholar] [CrossRef]
- Mach, N.; Midoux, C.; Leclercq, S.; Pennarun, S.; Le Moyec, L.; Rué, O.; Barrey, E. Mining the equine gut metagenome: Poorly-characterized taxa associated with cardiovascular fitness in endurance athletes. Commun. Biol. 2022, 5, 1032. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sävilammi, T.; Alakangas, R.-R.; Häyrynen, T.; Uusi-Heikkilä, S. Gut Microbiota Profiling as a Promising Tool to Detect Equine Inflammatory Bowel Disease (IBD). Animals 2024, 14, 2396. https://doi.org/10.3390/ani14162396
Sävilammi T, Alakangas R-R, Häyrynen T, Uusi-Heikkilä S. Gut Microbiota Profiling as a Promising Tool to Detect Equine Inflammatory Bowel Disease (IBD). Animals. 2024; 14(16):2396. https://doi.org/10.3390/ani14162396
Chicago/Turabian StyleSävilammi, Tiina, Rinna-Riikka Alakangas, Tuomas Häyrynen, and Silva Uusi-Heikkilä. 2024. "Gut Microbiota Profiling as a Promising Tool to Detect Equine Inflammatory Bowel Disease (IBD)" Animals 14, no. 16: 2396. https://doi.org/10.3390/ani14162396
APA StyleSävilammi, T., Alakangas, R.-R., Häyrynen, T., & Uusi-Heikkilä, S. (2024). Gut Microbiota Profiling as a Promising Tool to Detect Equine Inflammatory Bowel Disease (IBD). Animals, 14(16), 2396. https://doi.org/10.3390/ani14162396