

Genomic Characterization of Local Croatian Sheep Breeds-Effective Population Size, Inbreeding & Signatures of Selection

 ,
,  ,
, 
 , ,
, ,  , ,
, ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and SNP Genotyping
2.2. Runs of Homozygosity
2.3. Estimation of GenomicCoefficients of Inbreeding and Analysis of Signatures of Selection
3. Results
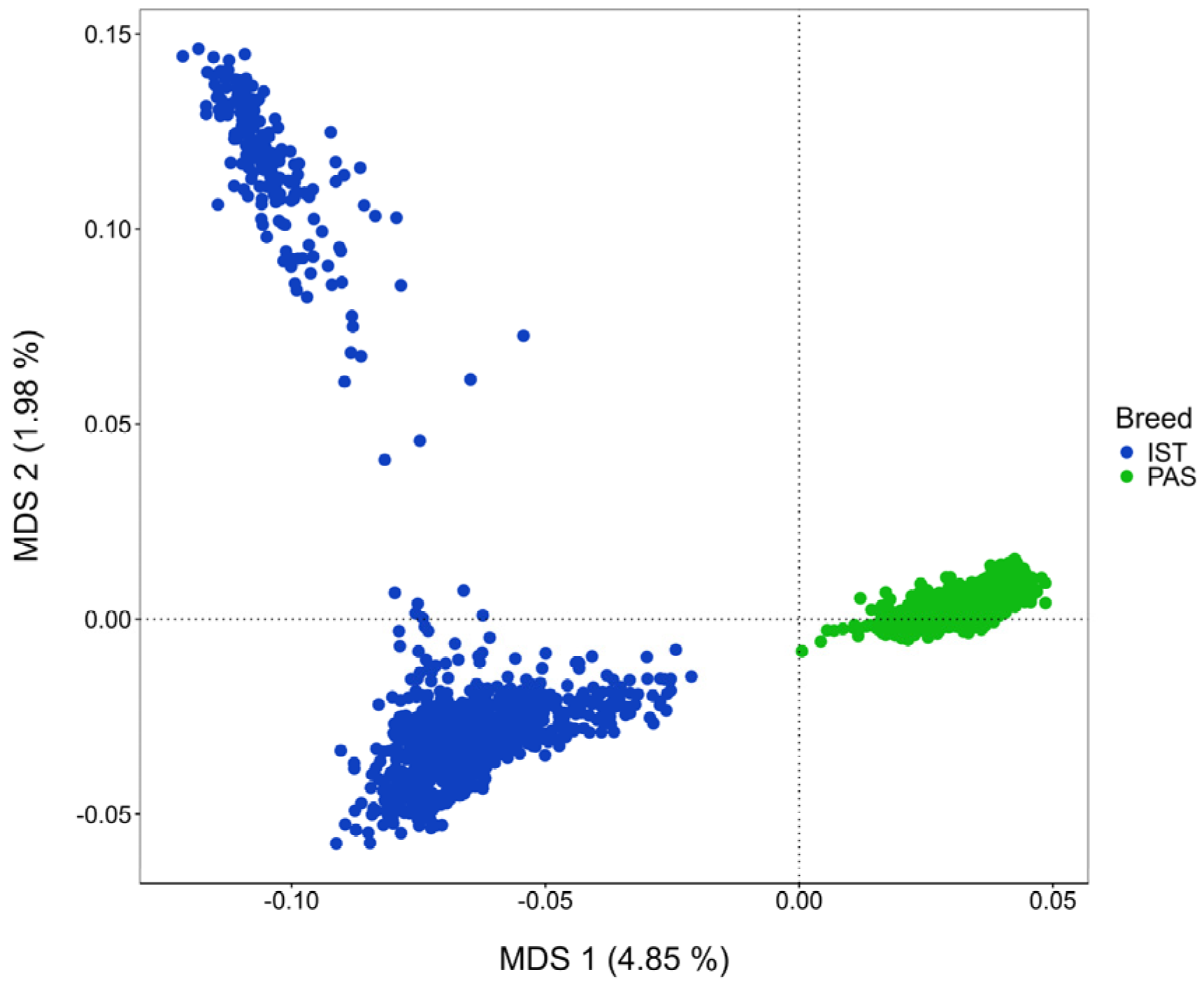
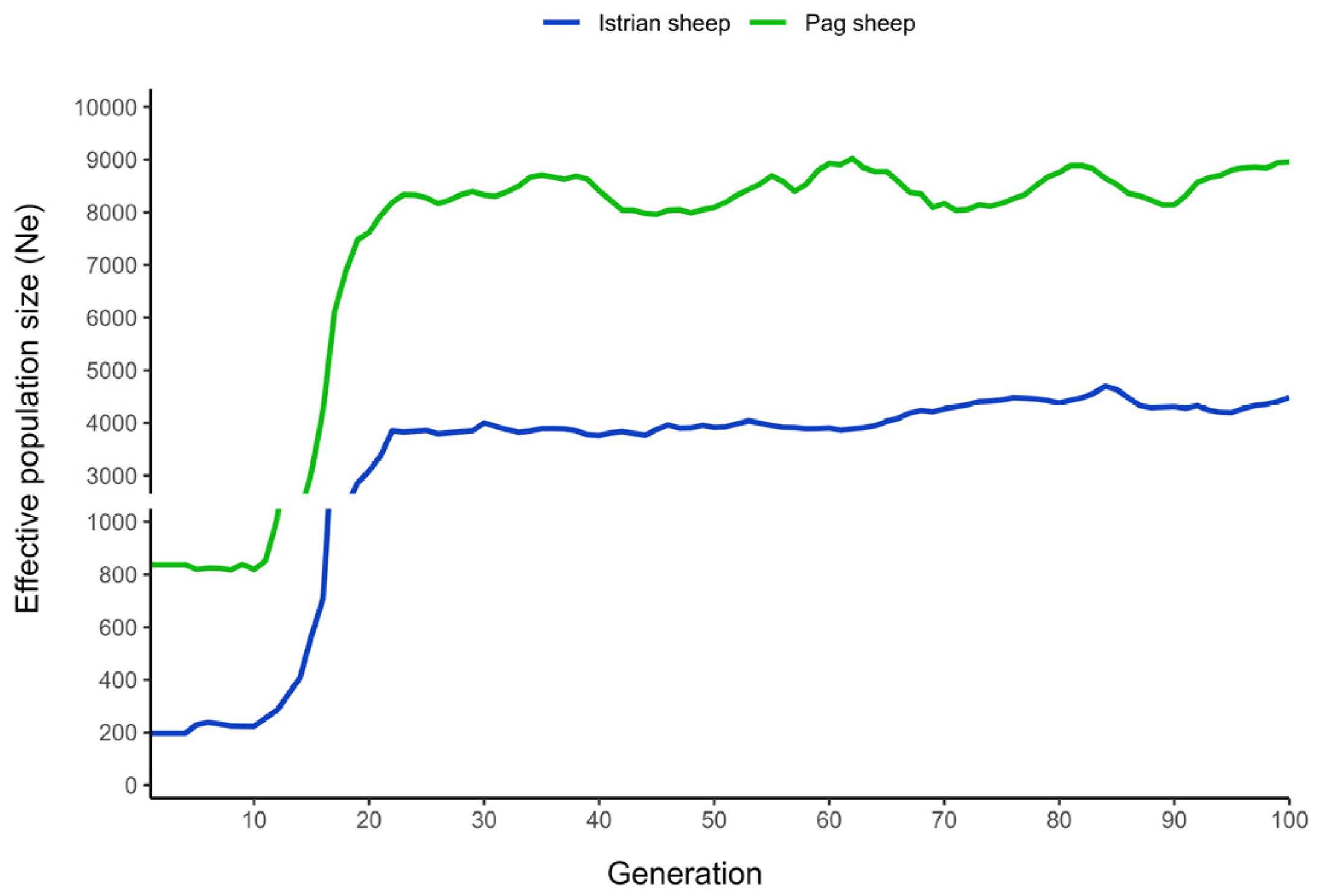
3.1. Genetic Relationship and Effective Population Size
3.2. Genomic Inbreeding
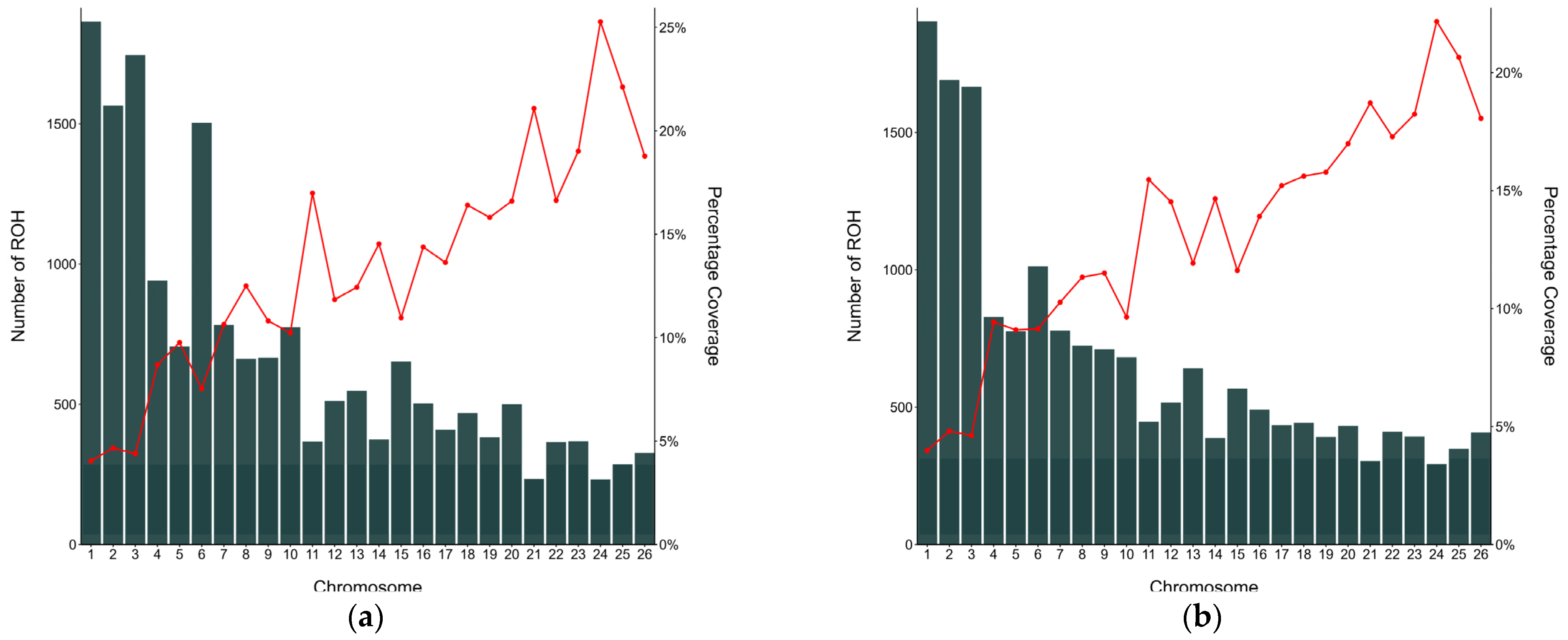
3.3. Characterisation of Runs of Homozygosity and Runs of Homozygosity Island
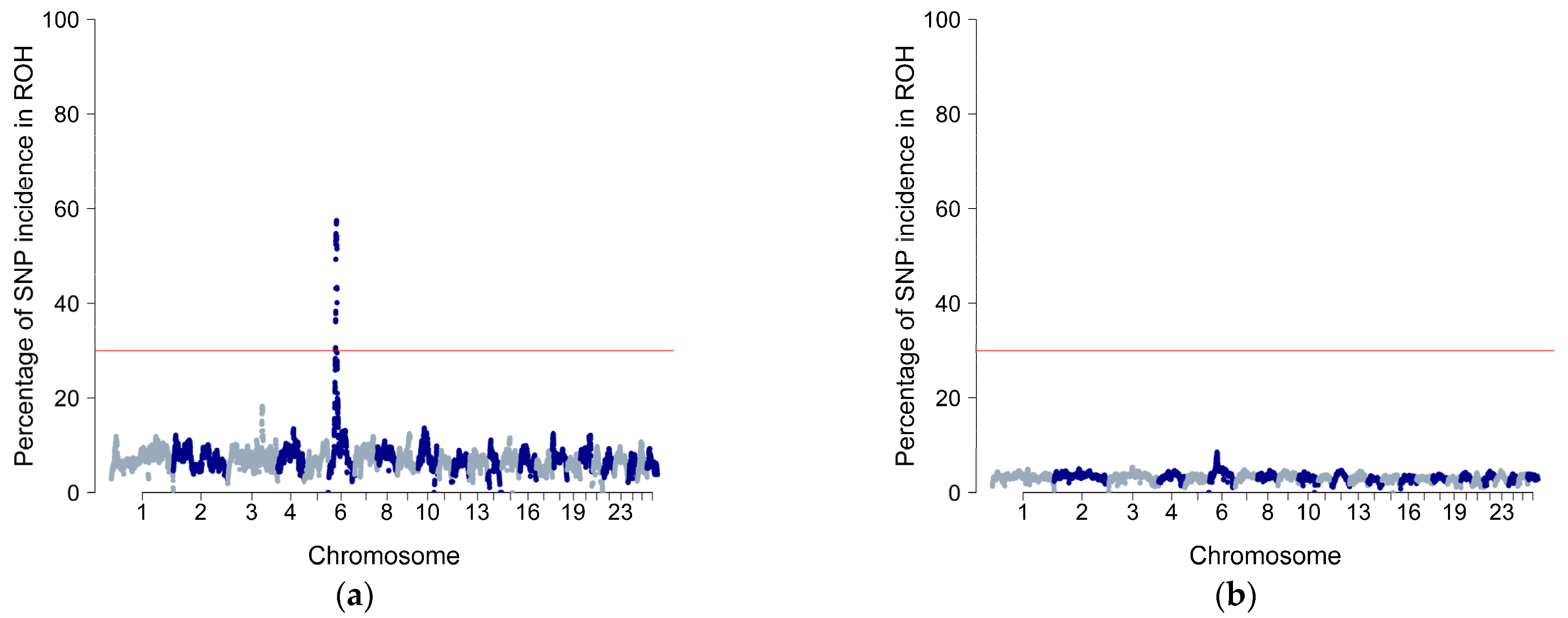
3.4. Gene and QTL Annotation
4. Discussion
4.1. Effective Population Size, Runs of Homozygosity and Genomic Inbreeding
4.2. Selection Signatures and Functional Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. The State of the World’s Animal Genetic Resources for Food and Agriculture; FAO Commission on Genetic Resources for Food and Agriculture Assessments: Rome, Italy, 2007; ISBN 978-92-5-105762-9. [Google Scholar]
- FAO. The Second Report on the State of the World’s Animal Genetic Resources for Food and Agriculture; FAO Commission on Genetic Resources for Food and Agriculture Assessments: Rome, Italy, 2015; ISBN 978-92-5-108820-3. [Google Scholar]
- Groeneveld, L.F.; Lenstra, J.A.; Eding, H.; Toro, M.A.; Scherf, B.; Pilling, D.; Negrini, R.; Finlay, E.K.; Jianlin, H.; Groeneveld, E.; et al. Genetic Diversity in Farm Animals—A Review. Anim. Genet. 2010, 41, 6–31. [Google Scholar] [CrossRef]
- Biscarini, F.; Nicolazzi, E.L.; Stella, A.; Boettcher, P.J.; Gandini, G. Challenges and Opportunities in Genetic Improvement of Local Livestock Breeds. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Mioč, B.; Prpić, Z.; Barać, Z.; Vnučec, I. Istarska ovca hrvatska izvorna pasmina; Hrvatski savez uzgajivača ovaca i koza: Zagreb, Croatia, 2012; ISBN 978-953-56869-1-0. [Google Scholar]
- CAAF. Annual report for year 2023; Croatian Agency for Agriculture and Food: Osijek, Croatia, 2023.
- European Regional Focal Point. Animal Genetic Resources Strategy for Europe; European Regional Focal Point: Paris, France, 2022; p. 38. [Google Scholar]
- Wellmann, R.; Hartwig, S.; Bennewitz, J. Optimum Contribution Selection for Conserved Populations with Historic Migration. Genet. Sel. Evol. 2012, 44, 34. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Drzaic, I.; Curik, I.; Lukic, B.; Shihabi, M.; Li, M.-H.; Kantanen, J.; Mastrangelo, S.; Ciani, E.; Lenstra, J.A.; Cubric-Curik, V. High-Density Genomic Characterization of Native Croatian Sheep Breeds. Front. Genet. 2022, 13, 940736. [Google Scholar] [CrossRef] [PubMed]
- Chessari, G.; Criscione, A.; Tolone, M.; Bordonaro, S.; Rizzuto, I.; Riggio, S.; Macaluso, V.; Moscarelli, A.; Portolano, B.; Sardina, M.T.; et al. High-Density SNP Markers Elucidate the Genetic Divergence and Population Structure of Noticiana Sheep Breed in the Mediterranean Context. Front. Vet. Sci. 2023, 10, 1127354. [Google Scholar] [CrossRef]
- Mioč, B.; Pavić, V.; Barać, Z.; Vnučec, I.; Prpić, Z.; Mulc, D.; Špehar, M. PROGRAM UZGOJA OVACA U REPUBLICI HRVATSKOJ (National Breeding and Selection Programme for Sheep); Hrvatski savez uzgajivača ovaca i koza: Zagreb, Croatia, 2011. [Google Scholar]
- ICAR. International Committee for Animal Recording Section 16. Guidelines for Performance Recording in Dairy Sheep and Dairy Goats; ICAR: New Delhi, India; p. 2018.
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Santiago, E.; Novo, I.; Pardiñas, A.F.; Saura, M.; Wang, J.; Caballero, A. Recent Demographic History Inferred by High-Resolution Analysis of Linkage Disequilibrium. Mol. Biol. Evol. 2020, 37, 3642–3653. [Google Scholar] [CrossRef]
- Milanesi, M.; Capomaccio, S.; Vajana, E.; Bomba, L.; Garcia, J.F.; Ajmone-Marsan, P.; Colli, L. BITE: An R Package for Biodiversity Analyses. BioRxiv 2017. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to Study Runs of Homozygosity Using PLINK? A Guide for Analyzing Medium Density SNP Data in Livestock and Pet Species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating Autozygosity from High-Throughput Information: Effects of SNP Density and Genotyping Errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of Homozygosity Reveal Highly Penetrant Recessive Loci in Schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of Homozygosity and Population History in Cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Gorssen, W.; Meyermans, R.; Buys, N.; Janssens, S. SNP Genotypes Reveal Breed Substructure, Selection Signatures and Highly Inbred Regions in Piétrain Pigs. Anim. Genet. 2020, 51, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Gorssen, W.; Meyermans, R.; Janssens, S.; Buys, N. A Publicly Available Repository of ROH Islands Reveals Signatures of Selection in Different Livestock and Pet Species. Genet. Sel. Evol. 2021, 53, 2. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The Distribution of Runs of Homozygosity and Selection Signatures in Six Commercial Meat Sheep Breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R Package for Genomic Annotation and Integration of Multiple Data Sources in Livestock for Positional Candidate Loci. GigaScience 2020, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Yurchenko, A.A.; Deniskova, T.E.; Yudin, N.S.; Dotsev, A.V.; Khamiruev, T.N.; Selionova, M.I.; Egorov, S.V.; Reyer, H.; Wimmers, K.; Brem, G.; et al. High-Density Genotyping Reveals Signatures of Selection Related to Acclimation and Economically Important Traits in 15 Local Sheep Breeds from Russia. BMC Genom. 2019, 20, 294. [Google Scholar] [CrossRef] [PubMed]
- La, Y.; Zhang, X.; Li, F.; Zhang, D.; Li, C.; Mo, F.; Wang, W. Molecular Characterization and Expression of SPP1, LAP3 and LCORL and Their Association with Growth Traits in Sheep. Genes 2019, 10, 616. [Google Scholar] [CrossRef] [PubMed]
- Al Kalaldeh, M.; Gibson, J.; Lee, S.H.; Gondro, C.; Van Der Werf, J.H.J. Detection of Genomic Regions Underlying Resistance to Gastrointestinal Parasites in Australian Sheep. Genet. Sel. Evol. 2019, 51, 37. [Google Scholar] [CrossRef] [PubMed]
- Kasap, A.; Ramljak, J.; Špehar, M. Estimation of Population-Specific Genetic Parameters Important for Long-Term Optimum Contribution Selection—Case Study on a Dairy Istrian Sheep Breed. Animals 2021, 11, 2356. [Google Scholar] [CrossRef] [PubMed]
- Kasap, A.; Mijadžiković, Z.; Smutni, B.; Mioč, B.; Špehar, M. Estimation of Inbreeding Coefficient and Generation Interval in Pag Sheep – Baseline for Development of Optimum Contribution Selection. In Proceedings of the 58th Croatian & 18th International Symposium on Agriculture, Dubrovnik, Croatia, 15 February 2023; University of Zagreb Faculty of Agriculture: Dubrovnik, Croatia, 2023; pp. 320–325. [Google Scholar]
- Pajalić, J. Ovčarenje i Ovca Na Otoku Pagu. Poljopr. Znan. Smotra 1953, 15, 279–313. [Google Scholar]
- Meuwissen, T. Genetic Management of Small Populations: A Review. Acta Agric. Scand. Sect. A—Anim. Sci. 2009, 59, 71–79. [Google Scholar] [CrossRef]
- Špehar, M.; Ramljak, J.; Kasap, A. Estimation of Genetic Parameters and the Effect of Inbreeding on Dairy Traits in Istrian Sheep. Ital. J. Anim. Sci. 2022, 21, 331–342. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-Wide Scan for Runs of Homozygosity Identifies Potential Candidate Genes Associated with Local Adaptation in Valle Del Belice Sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Ciani, E.; Sardina, M.T.; Sottile, G.; Pilla, F.; Portolano, B.; the Bi.Ov. Ita Consortium. Runs of Homozygosity Reveal Genome-wide Autozygosity in Italian Sheep Breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef]
- Gurgul, A.; Jasielczuk, I.; Miksza-Cybulska, A.; Kawęcka, A.; Szmatoła, T.; Krupiński, J. Evaluation of Genetic Differentiation and Genome-Wide Selection Signatures in Polish Local Sheep Breeds. Livest. Sci. 2021, 251, 104635. [Google Scholar] [CrossRef]
- Lukic, B.; Curik, I.; Drzaic, I.; Galić, V.; Shihabi, M.; Vostry, L.; Cubric-Curik, V. Genomic Signatures of Selection, Local Adaptation and Production Type Characterisation of East Adriatic Sheep Breeds. J. Anim. Sci. Biotechnol. 2023, 14, 142. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Cao, J.; Wu, M.; Ma, X.; Liu, Z.; Liu, R.; Zhao, F.; Wei, C.; Du, L. Genome-Wide Specific Selection in Three Domestic Sheep Breeds. PLoS ONE 2015, 10, e0128688. [Google Scholar] [CrossRef]
- Abo-Ismail, M.K.; Lansink, N.; Akanno, E.; Karisa, B.K.; Crowley, J.J.; Moore, S.S.; Bork, E.; Stothard, P.; Basarab, J.A.; Plastow, G.S. Development and Validation of a Small SNP Panel for Feed Efficiency in Beef Cattle1. J. Anim. Sci. 2018, 96, 375–397. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, S.; Zhang, H.; Zhang, Z.; Chen, N.; Li, Z.; Sun, H.; Liu, X.; Lyu, S.; Wang, X.; et al. Assessing Genomic Diversity and Signatures of Selection in Jiaxian Red Cattle Using Whole-Genome Sequencing Data. BMC Genom. 2021, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Ramón, M.; Calvo, J.H.; Jiménez, M.Á.; Freire, F.; Vázquez, J.M.; Arranz, J.J. Genome-Wide Association Studies for Sperm Traits in Assaf Sheep Breed. Animal 2021, 15, 100065. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Reverter, A.; Prayaga, K.C.; Chan, E.K.F.; Johnston, D.J.; Hawken, R.J.; Fordyce, G.; Garcia, J.F.; Sonstegard, T.S.; Bolormaa, S.; et al. The Genetic Architecture of Climatic Adaptation of Tropical Cattle. PLoS ONE 2014, 9, e113284. [Google Scholar] [CrossRef] [PubMed]
- Naserkheil, M.; Bahrami, A.; Lee, D.; Mehrban, H. Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle. Animals 2020, 10, 1836. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, J.; Sun, D.; Ma, P.; Ding, X.; Yu, Y.; Zhang, Q. Genome Wide Association Studies for Milk Production Traits in Chinese Holstein Population. PLoS ONE 2010, 5, e13661. [Google Scholar] [CrossRef]
- Zaitoun, I.; Khatib, H. Assessment of Genomic Imprinting of SLC38A4, NNAT, NAP1L5, and H19 in Cattle. BMC Genet. 2006, 7, 49. [Google Scholar] [CrossRef]
- Wei, C.; Wang, H.; Liu, G.; Wu, M.; Cao, J.; Liu, Z.; Liu, R.; Zhao, F.; Zhang, L.; Lu, J.; et al. Genome-Wide Analysis Reveals Population Structure and Selection in Chinese Indigenous Sheep Breeds. BMC Genom. 2015, 16, 194. [Google Scholar] [CrossRef]
- Van der Steen, H.; Southwood, O.; Vries, A.; Short, T.; McLaren, D.; Wei, M.; Plastow, G. Evidence of a New Genetic Marker for Litter Size in Meishan Synthetic Pigs. In Proceedings of International Conference on Animal Biotechnology; International Academic Publishers: Beijing, China, 1997; pp. 17–20. [Google Scholar]
- Weintraub, A.S.; Lin, X.; Itskovich, V.V.; Aguinaldo, J.G.S.; Chaplin, W.F.; Denhardt, D.T.; Fayad, Z.A. Prenatal Detection of Embryo Resorption in Osteopontin-Deficient Mice Using Serial Noninvasive Magnetic Resonance Microscopy. Pediatr. Res. 2004, 55, 419–424. [Google Scholar] [CrossRef]
- Rangaswami, H.; Bulbule, A.; Kundu, G.C. Osteopontin: Role in Cell Signaling and Cancer Progression. Trends Cell Biol. 2006, 16, 79–87. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Liao, W.-L.; Wang, C.-H.; Tsai, L.-P.; Tang, C.-H.; Chen, C.-H.; Wu, J.-Y.; Liang, W.-M.; Hsieh, A.-R.; Cheng, C.-F.; et al. Association of Human Height-Related Genetic Variants with Familial Short Stature in Han Chinese in Taiwan. Sci. Rep. 2017, 7, 6372. [Google Scholar] [CrossRef]
- Saif, R.; Henkel, J.; Jagannathan, V.; Drögemüller, C.; Flury, C.; Leeb, T. The LCORL Locus Is under Selection in Large-Sized Pakistani Goat Breeds. Genes 2020, 11, 168. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Dall’Olio, S.; Nanni Costa, L.; Tinarelli, S.; Gallo, M.; Fontanesi, L. Runs of Homozygosity Islands in Italian Cosmopolitan and Autochthonous Pig Breeds Identify Selection Signatures in the Porcine Genome. Livest. Sci. 2020, 240, 104219. [Google Scholar] [CrossRef]
- Ablondi, M.; Dadousis, C.; Vasini, M.; Eriksson, S.; Mikko, S.; Sabbioni, A. Genetic Diversity and Signatures of Selection in a Native Italian Horse Breed Based on SNP Data. Animals 2020, 10, 1005. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Di, J.; Han, B.; Chen, L.; Liu, M.; Li, W. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes Related to Economically Important Traits in Chinese Merino. Animals 2020, 10, 524. [Google Scholar] [CrossRef] [PubMed]
- Rochus, C.M.; Tortereau, F.; Plisson-Petit, F.; Restoux, G.; Moreno-Romieux, C.; Tosser-Klopp, G.; Servin, B. Revealing the Selection History of Adaptive Loci Using Genome-Wide Scans for Selection: An Example from Domestic Sheep. BMC Genom. 2018, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ramilo, S.T.; Reverter, A.; Legarra, A. Islands of Runs of Homozygosity Indicate Selection Signatures in Ovis Aries 6 (OAR6) of French Dairy Sheep. JDS Commun. 2021, 2, 132–136. [Google Scholar] [CrossRef]
- Signer-Hasler, H.; Flury, C.; Haase, B.; Burger, D.; Simianer, H.; Leeb, T.; Rieder, S. A Genome-Wide Association Study Reveals Loci Influencing Height and Other Conformation Traits in Horses. PLoS ONE 2012, 7, e37282. [Google Scholar] [CrossRef]
- Al-Mamun, H.A.; Kwan, P.; Clark, S.A.; Ferdosi, M.H.; Tellam, R.; Gondro, C. Genome-Wide Association Study of Body Weight in Australian Merino Sheep Reveals an Orthologous Region on OAR6 to Human and Bovine Genomic Regions Affecting Height and Weight. Genet. Sel. Evol. 2015, 47, 66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bai, C.; Shi, L.; He, Y.; Hu, M.; Sun, H.; Peng, H.; Lai, W.; Jiao, S.; Zhao, Z.; et al. Detection of Selection Signatures in South African Mutton Merino Sheep Using Whole-Genome Sequencing Data. Anim. Genet. 2022, 53, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Matika, O.; Riggio, V.; Anselme-Moizan, M.; Law, A.S.; Pong-Wong, R.; Archibald, A.L.; Bishop, S.C. Genome-Wide Association Reveals QTL for Growth, Bone and in Vivo Carcass Traits as Assessed by Computed Tomography in Scottish Blackface Lambs. Genet. Sel. Evol. 2016, 48, 11. [Google Scholar] [CrossRef] [PubMed]
- Zlobin, A.S.; Nikulin, P.S.; Volkova, N.A.; Zinovieva, N.A.; Iolchiev, B.S.; Bagirov, V.A.; Borodin, P.M.; Aksenovich, T.I.; Tsepilov, Y.A. Multivariate Analysis Identifies Eight Novel Loci Associated with Meat Productivity Traits in Sheep. Genes 2021, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Ghoreishifar, S.M.; Eriksson, S.; Johansson, A.M.; Khansefid, M.; Moghaddaszadeh-Ahrabi, S.; Parna, N.; Davoudi, P.; Javanmard, A. Signatures of Selection Reveal Candidate Genes Involved in Economic Traits and Cold Acclimation in Five Swedish Cattle Breeds. Genet. Sel. Evol. 2020, 52, 52. [Google Scholar] [CrossRef]
- Ramos, Z.; Garrick, D.J.; Blair, H.T.; De Barbieri, I.; Ciappesoni, G.; Montossi, F.; Kenyon, P.R. Genetic and Phenotypic Relationships between Ewe Reproductive Performance and Wool and Growth Traits in Uruguayan Ultrafine Merino Sheep. J. Anim. Sci. 2023, 101, skad071. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Istrian Sheep (IS) | Pag Sheep (PS) | ||
|---|---|---|---|
| nROH/animal | 13.05 | 5.94 | |
| ROH classes | ROH2–4 ROH4–8 ROH8–16 ROH>16 | 2320 (15%) 6683 (43%) 4067 (26%) 2506 (16%) | 2961 (19%) 6147 (39%) 3779 (24%) 2767 (18%) |
| Inbreeding coefficients /classes (%) | FHOM | 5.80 | 3.90 |
| FROH>2 | 6.24 | 2.88 | |
| FROH2–4 | 0.26 | 0.22 | |
| FROH4–8 | 1.65 | 0.61 | |
| FROH8–16 | 2.14 | 0.75 | |
| FROH>16 | 3.72 | 1.58 | |
| Chr | Start (bp) | End (bp) | Length (bp) | Gene | Gene_ID | Description | Functions |
|---|---|---|---|---|---|---|---|
| 6 | 33,699,466 | 35,188,219 | 1,488,754 | CCSER1 | ENSOARG 00000007488 | Coiled-coil serine rich protein 1 | Growth, feed intake, reproduction |
| 6 | 36,709,616 | 36,855,827 | 146,212 | HERC3 | ENSOARG 00020023461 | HECT and RLD domain containing E3 ubiquitin protein ligase 3 | Growth, immune responses |
| 6 | 36,739,315 | 36,741,723 | 2409 | NAP1L5 | ENSOARG 00020037274 | Nucleosome assembly protein 1 like 5 | Growth and development (chromatin organization) |
| 6 | 36,902,696 | 36,990,169 | 87,474 | PYURF | ENSOARG 00020035580 | PIGY-phosphatidylinositol glycan anchor biosynthesis class Y | Growth, immune response |
| 6 | 37,282,161 | 37,348,326 | 66,166 | PKD2 | ENSOARG 00020024032 | Polycystin 2, transient receptor potential cation channel | Growth, milk, feed intake |
| 6 | 37,369,583 | 37,425,206 | 55,624 | SPP1 | ENSOARG 00020024091 | Secreted phosphoprotein 1 (osteopontin) | Growth, milk, immune responses |
| 6 | 38,052,622 | 38,222,929 | 170,308 | LCORL | ENSOARG 00020024486 | Ligand dependent nuclear receptor corepressor like | Growth |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramljak, J.; Špehar, M.; Ceranac, D.; Držaić, V.; Pocrnić, I.; Barać, D.; Mioč, B.; Širić, I.; Barać, Z.; Ivanković, A.; et al. Genomic Characterization of Local Croatian Sheep Breeds-Effective Population Size, Inbreeding & Signatures of Selection. Animals 2024, 14, 1928. https://doi.org/10.3390/ani14131928
Ramljak J, Špehar M, Ceranac D, Držaić V, Pocrnić I, Barać D, Mioč B, Širić I, Barać Z, Ivanković A, et al. Genomic Characterization of Local Croatian Sheep Breeds-Effective Population Size, Inbreeding & Signatures of Selection. Animals. 2024; 14(13):1928. https://doi.org/10.3390/ani14131928
Chicago/Turabian StyleRamljak, Jelena, Marija Špehar, Dora Ceranac, Valentino Držaić, Ivan Pocrnić, Dolores Barać, Boro Mioč, Ivan Širić, Zdravko Barać, Ante Ivanković, and et al. 2024. "Genomic Characterization of Local Croatian Sheep Breeds-Effective Population Size, Inbreeding & Signatures of Selection" Animals 14, no. 13: 1928. https://doi.org/10.3390/ani14131928
APA StyleRamljak, J., Špehar, M., Ceranac, D., Držaić, V., Pocrnić, I., Barać, D., Mioč, B., Širić, I., Barać, Z., Ivanković, A., & Kasap, A. (2024). Genomic Characterization of Local Croatian Sheep Breeds-Effective Population Size, Inbreeding & Signatures of Selection. Animals, 14(13), 1928. https://doi.org/10.3390/ani14131928