
Genome Evolution and the Future of Phylogenomics of Non-Avian Reptiles

Simple Summary
Abstract
1. Introduction
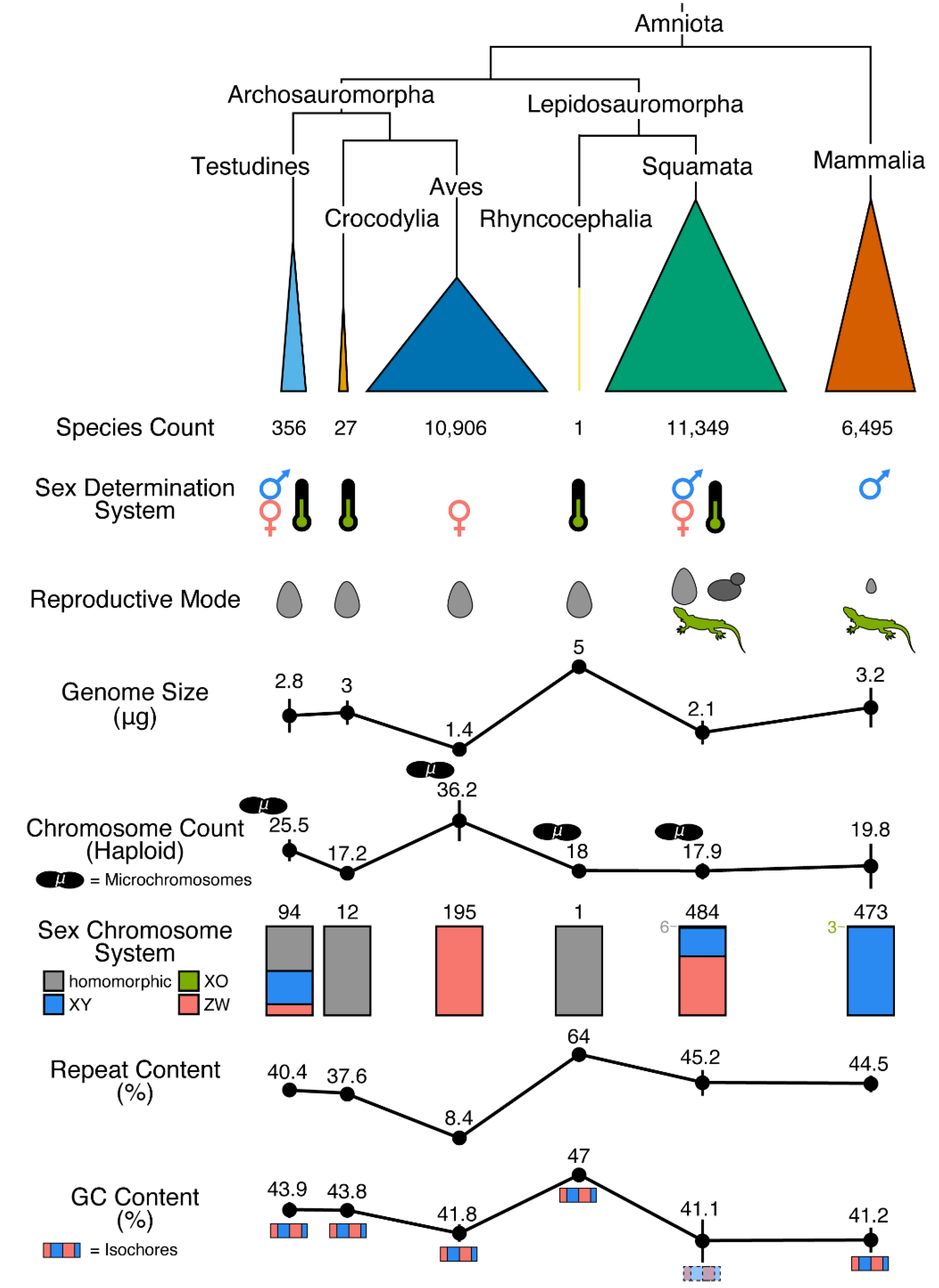
2. Non-Avian Reptiles Are Highly Variable in Physical Traits with Strong Links to the Genome
3. Substantial Variation in Genome Size and Karyotype among Non-Avian Reptiles
4. Dynamic Features of Sequence Composition in Non-Avian Reptiles
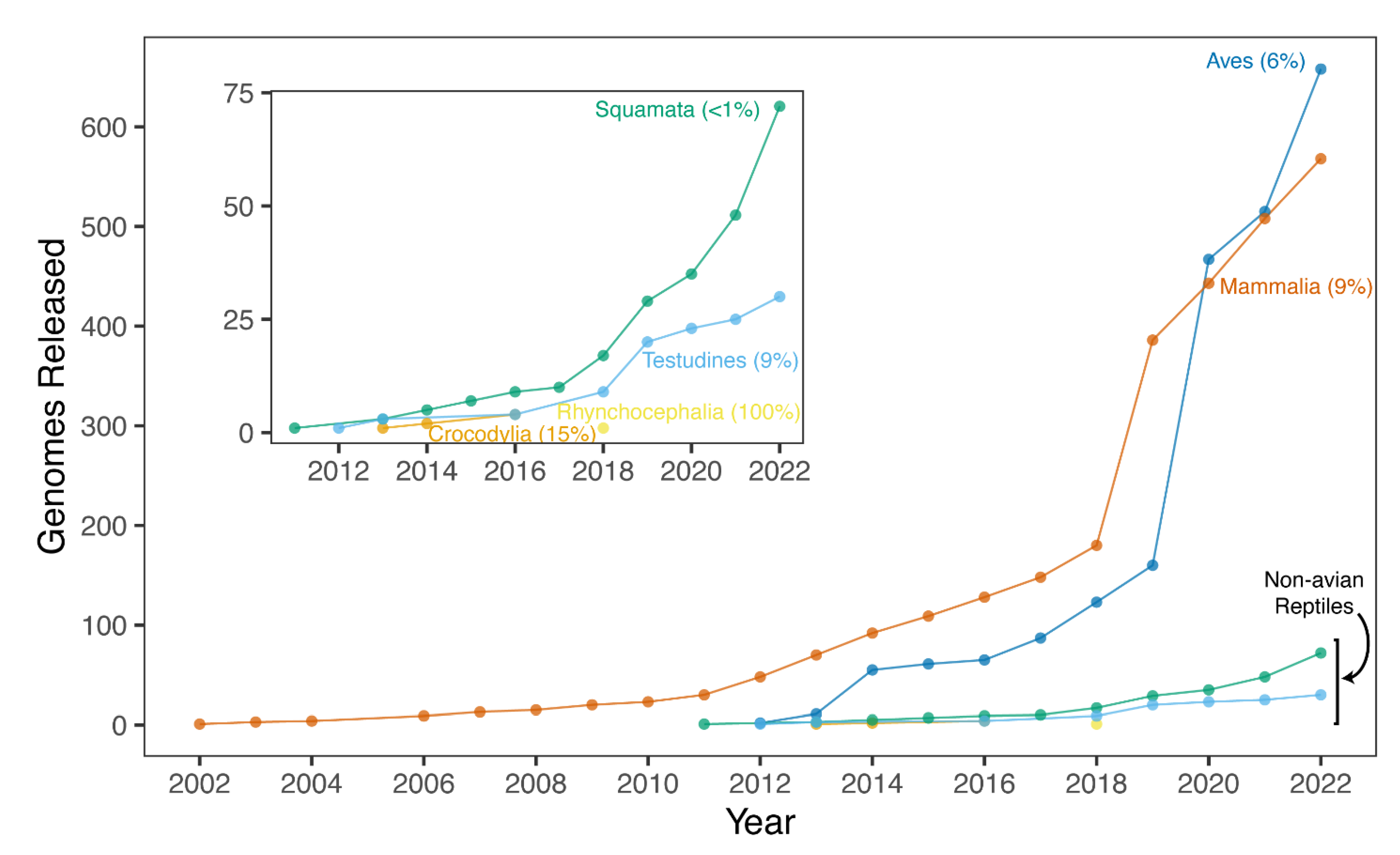
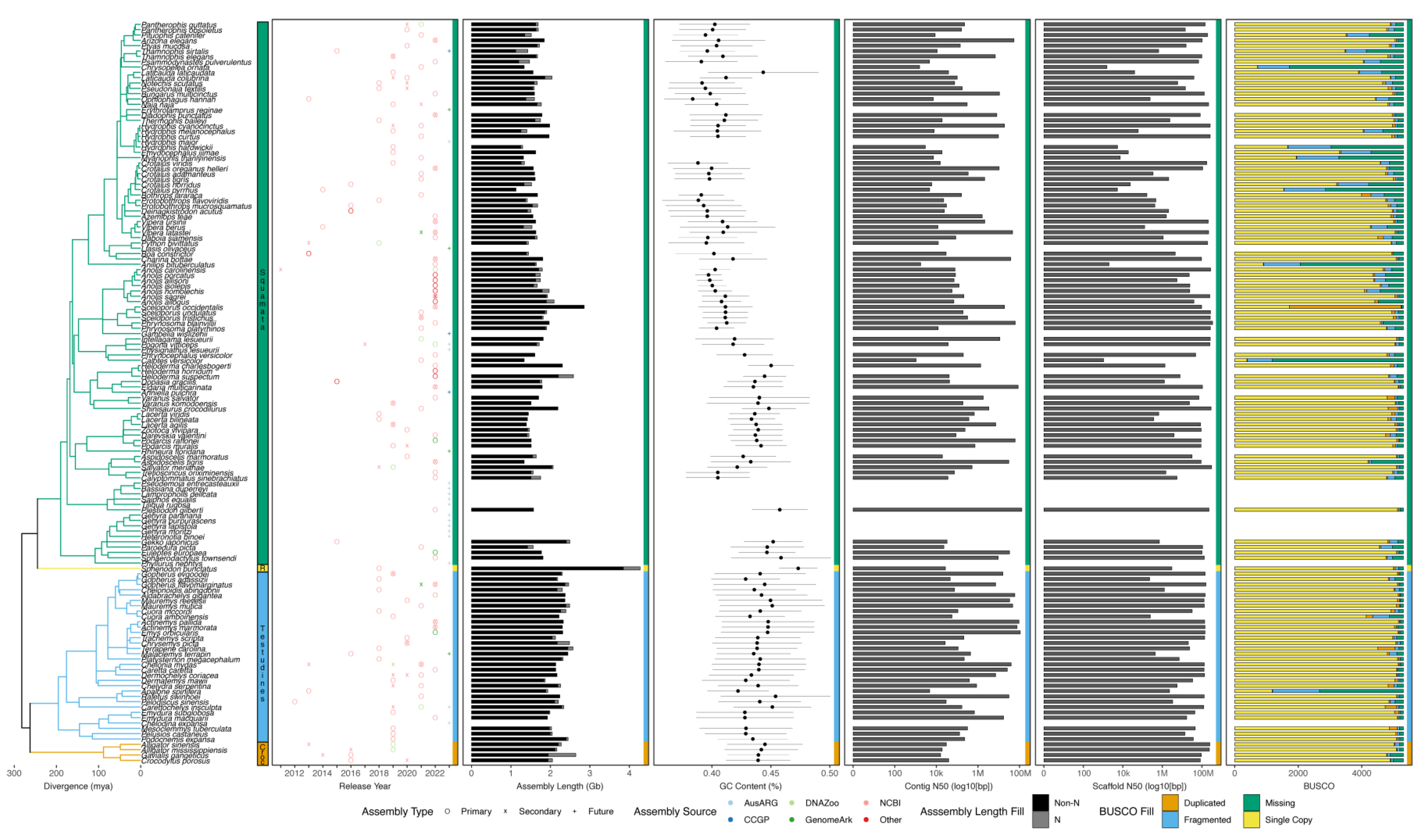
5. Summary of Available Reference Genomes for Non-Avian Reptiles
6. Why Are There So Few Genomes of Non-Avian Reptiles?
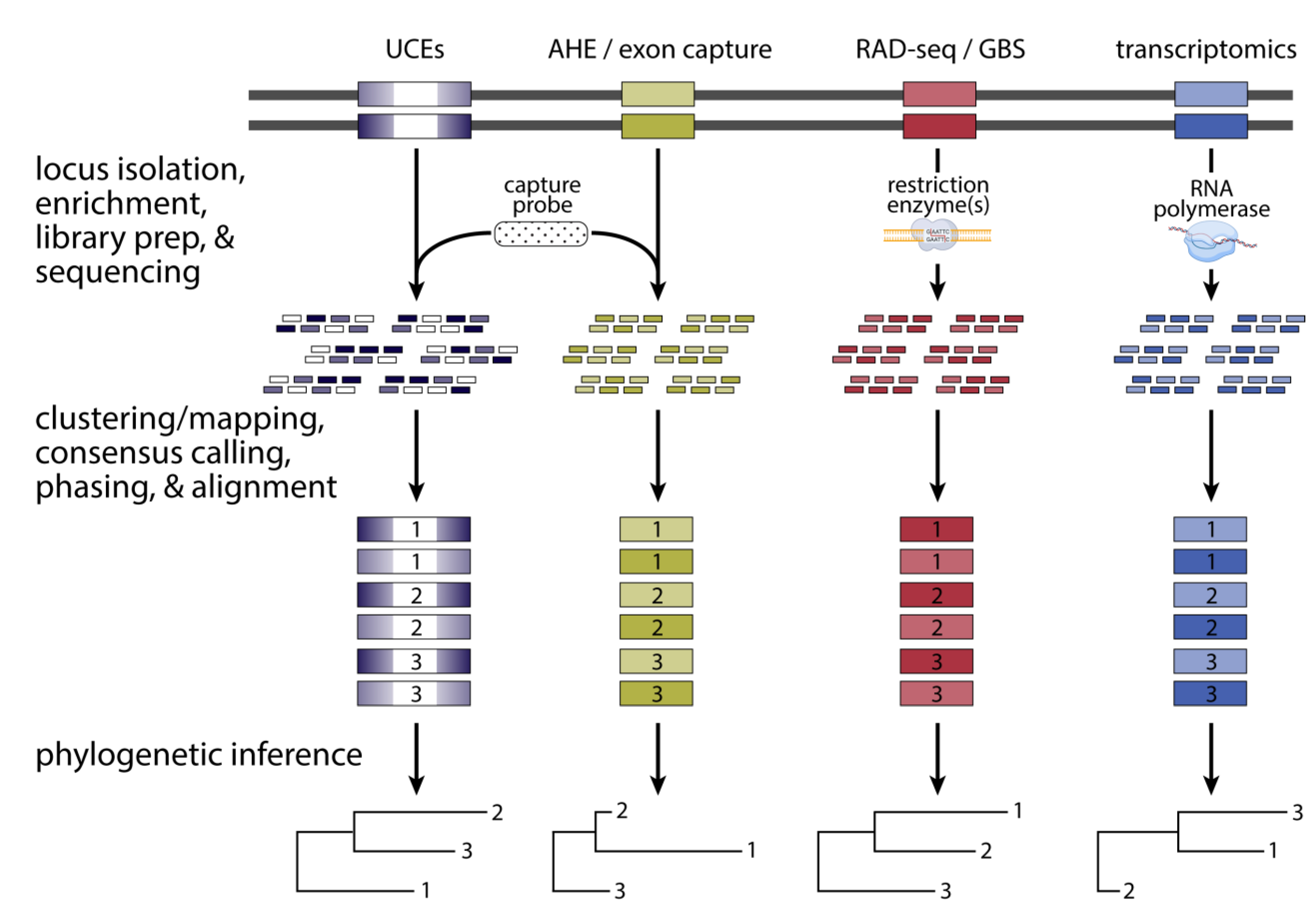
7. First-Generation Phylogenomic Data Acquisition: Reduced Representation Approaches
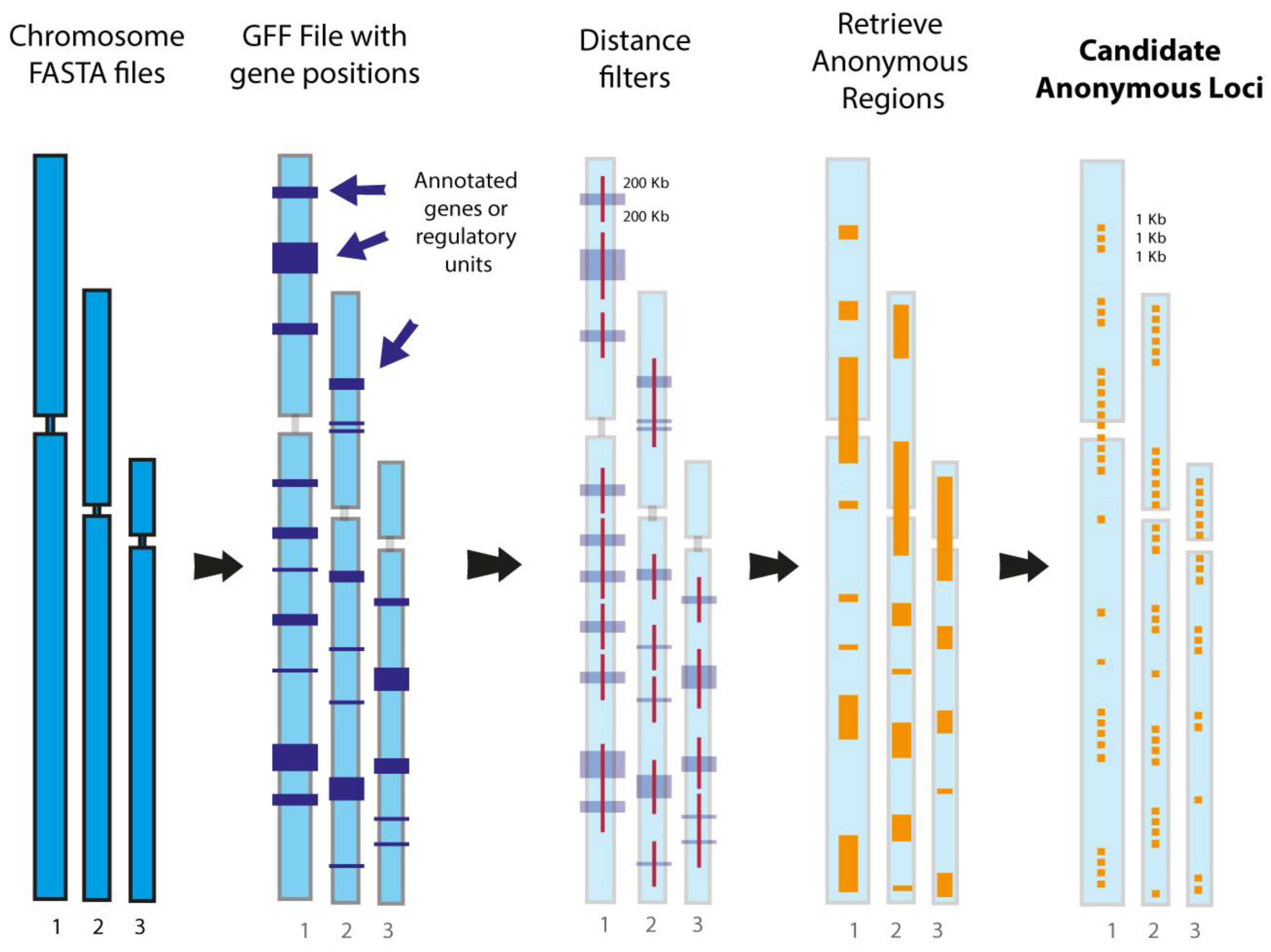
8. Genome-Scale Phylogenomics: In Silico Investigation of Markers Extracted from Whole Genomes
9. Allele Phasing Is a Much-Neglected Component of Most Phylogenomic Workflows
10. New Reptile Genomes Will Fuel the Future of Reptile Phylogenomics and Genome-Phenotype Discovery via Comparative Genomics
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amniote Paleobiology: Perspectives on the Evolution of Mammals, Birds, and Reptiles; Carrano, M.T., Gaudin, T.J., Blob, R.W., Wible, J.R., Eds.; University of Chicago Press: Chicago, IL, USA, 2006; ISBN 978-0-226-09478-6. [Google Scholar]
- Shedlock, A.M.; Edwards, S.V. Amniotes (Amniota). In The Timetree of Life; Hedges, S.B., Kumar, S., Eds.; Oxford University Press: New York, NY, USA, 2009; pp. 375–379. ISBN 978-0-19-953503-3. [Google Scholar]
- Sues, H.-D. The Rise of Reptiles: 320 Million Years of Evolution, Illustrated ed.; Johns Hopkins University Press: Baltimore, MA, USA, 2019; ISBN 978-1-4214-2867-3. [Google Scholar]
- Hedges, S.B.; Dudley, J.; Kumar, S. TimeTree: A Public Knowledge-Base of Divergence Times among Organisms. Bioinformatics 2006, 22, 2971–2972. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Uetz, P.; Etzold, T. The EMBL/EBI Reptile Database. Herpetol. Rev. 1996, 27, 174–175. [Google Scholar]
- Uetz, P.; Freed, P.; Aguilar, R.; Reyes, F.; Hošek, J. The Reptile Database. 2022. Available online: http://www.reptile-database.org (accessed on 15 December 2022).
- Stöck, M.; Kratochvíl, L.; Kuhl, H.; Rovatsos, M.; Evans, B.J.; Suh, A.; Valenzuela, N.; Veyrunes, F.; Zhou, Q.; Gamble, T.; et al. A Brief Review of Vertebrate Sex Evolution with a Pledge for Integrative Research: Towards ‘Sexomics’. Philos. Trans. R. Soc. B Biol. Sci. 2021, 376, 20200426. [Google Scholar] [CrossRef]
- Capel, B. Vertebrate Sex Determination: Evolutionary Plasticity of a Fundamental Switch. Nat. Rev. Genet. 2017, 18, 675–689. [Google Scholar] [CrossRef]
- Angelini, F.; Ghiara, G. Reproductive Modes and Strategies in Vertebrate Evolution. Boll. Zool. 1984, 51, 121–203. [Google Scholar] [CrossRef]
- Avise, J.C. Evolutionary Perspectives on Clonal Reproduction in Vertebrate Animals. Proc. Natl. Acad. Sci. USA 2015, 112, 8867–8873. [Google Scholar] [CrossRef] [PubMed]
- Neaves, W.B.; Baumann, P. Unisexual Reproduction among Vertebrates. Trends Genet. 2011, 27, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R.; Nicol, J.A.; Tamm, H.; Kullman, B.; Kullman, K.; Leitch, I.J.; Murray, B.G.; Kapraun, D.F.; Greilhuber, J.; Bennett, M.D. Eukaryotic Genome Size Databases. Nucleic Acids Res. 2007, 35, D332–D338. [Google Scholar] [CrossRef]
- Ashman, T.-L.; Bachtrog, D.; Blackmon, H.; Goldberg, E.E.; Hahn, M.W.; Kirkpatrick, M.; Kitano, J.; Mank, J.E.; Mayrose, I.; Ming, R.; et al. Tree of Sex: A Database of Sexual Systems. Sci. Data 2014, 1, 140015. [Google Scholar] [CrossRef]
- Pasquesi, G.I.M.; Adams, R.H.; Card, D.C.; Schield, D.R.; Corbin, A.B.; Perry, B.W.; Reyes-Velasco, J.; Ruggiero, R.P.; Vandewege, M.W.; Shortt, J.A.; et al. Squamate Reptiles Challenge Paradigms of Genomic Repeat Element Evolution Set by Birds and Mammals. Nat. Commun. 2018, 9, 2774. [Google Scholar] [CrossRef]
- Green, R.E.; Braun, E.L.; Armstrong, J.; Earl, D.; Nguyen, N.; Hickey, G.; Vandewege, M.W.; St. John, J.A.; Capella-Gutiérrez, S.; Castoe, T.A.; et al. Three Crocodilian Genomes Reveal Ancestral Patterns of Evolution among Archosaurs. Science 2014, 346, 1254449. [Google Scholar] [CrossRef] [PubMed]
- Brian Simison, W.; Parham, J.F.; Papenfuss, T.J.; Lam, A.W.; Henderson, J.B. An Annotated Chromosome-Level Reference Genome of the Red-Eared Slider Turtle (Trachemys scripta elegans). Genome Biol. Evol. 2020, 12, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, N.J.; Rutherford, K.; Prost, S.; Tollis, M.; Winter, D.; Macey, J.R.; Adelson, D.L.; Suh, A.; Bertozzi, T.; Grau, J.H.; et al. The Tuatara Genome Reveals Ancient Features of Amniote Evolution. Nature 2020, 584, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of Genome Size Evolution in Birds and Mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef]
- Cao, D.; Wang, M.; Ge, Y.; Gong, S. Draft Genome of the Big-Headed Turtle Platysternon megacephalum. Sci. Data 2019, 6, 60. [Google Scholar] [CrossRef]
- Das, D.; Singh, S.K.; Bierstedt, J.; Erickson, A.; Galli, G.L.J.; Crossley, D.A., II; Rhen, T. Draft Genome of the Common Snapping Turtle, Chelydra serpentina, a Model for Phenotypic Plasticity in Reptiles. G3 Genes|Genomes|Genetics 2020, 10, 4299–4314. [Google Scholar] [CrossRef]
- Kitts, P.A.; Church, D.M.; Thibaud-Nissen, F.; Choi, J.; Hem, V.; Sapojnikov, V.; Smith, R.G.; Tatusova, T.; Xiang, C.; Zherikov, A.; et al. Assembly: A Resource for Assembled Genomes at NCBI. Nucleic Acids Res. 2016, 44, D73–D80. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Franklin, J.; Katsu, Y.; Iguchi, T.; Guillette, L.J.; Kimball, R.T.; Braun, E.L. Patterns of Vertebrate Isochore Evolution Revealed by Comparison of Expressed Mammalian, Avian, and Crocodilian Genes. J. Mol. Evol. 2007, 65, 259–266. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Braun, E.L. Turtle Isochore Structure Is Intermediate between Amphibians and Other Amniotes. Integr. Comp. Biol. 2008, 48, 454–462. [Google Scholar] [CrossRef]
- Costantini, M.; Clay, O.; Auletta, F.; Bernardi, G. An Isochore Map of Human Chromosomes. Genome Res. 2006, 16, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Costantini, M.; Filippo, M.D.; Auletta, F.; Bernardi, G. Isochore Pattern and Gene Distribution in the Chicken Genome. Gene 2007, 400, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.K.; Edwards, S.V.; Ponting, C.P. The Anolis Lizard Genome: An Amniote Genome without Isochores. Genome Biol. Evol. 2011, 3, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Costantini, M.; Greif, G.; Alvarez-Valin, F.; Bernardi, G. The Anolis Lizard Genome: An Amniote Genome without Isochores? Genome Biol. Evol. 2016, 8, 1048–1055. [Google Scholar] [CrossRef]
- St John, J.A.; Braun, E.L.; Isberg, S.R.; Miles, L.G.; Chong, A.Y.; Gongora, J.; Dalzell, P.; Moran, C.; Bed’Hom, B.; Abzhanov, A.; et al. Sequencing Three Crocodilian Genomes to Illuminate the Evolution of Archosaurs and Amniotes. Genome Biol. 2012, 13, 415. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, H.B.; Minx, P.; Warren, D.E.; Shedlock, A.M.; Thomson, R.C.; Valenzuela, N.; Abramyan, J.; Amemiya, C.T.; Badenhorst, D.; Biggar, K.K.; et al. The Western Painted Turtle Genome, a Model for the Evolution of Extreme Physiological Adaptations in a Slowly Evolving Lineage. Genome Biol. 2013, 14, R28. [Google Scholar] [CrossRef]
- Costantini, M.; Cammarano, R.; Bernardi, G. The Evolution of Isochore Patterns in Vertebrate Genomes. BMC Genom. 2009, 10, 146. [Google Scholar] [CrossRef]
- Janes, D.E.; Organ, C.L.; Fujita, M.K.; Shedlock, A.M.; Edwards, S.V. Genome Evolution in Reptilia, the Sister Group of Mammals. Annu. Rev. Genom. Hum. Genet. 2010, 11, 239–264. [Google Scholar] [CrossRef]
- Jennings, W.B. Phylogenomic Data Acquisition: Principles and Practice; CRC Press: Boca Raton, FL, USA, 2016; ISBN 978-1-315-18143-1. [Google Scholar]
- Duret, L.; Galtier, N. Biased Gene Conversion and the Evolution of Mammalian Genomic Landscapes. Annu. Rev. Genom. Hum. Genet. 2009, 10, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Romiguier, J.; Ranwez, V.; Douzery, E.J.P.; Galtier, N. Contrasting GC-Content Dynamics across 33 Mammalian Genomes: Relationship with Life-History Traits and Chromosome Sizes. Genome Res. 2010, 20, 1001–1009. [Google Scholar] [CrossRef]
- Figuet, E.; Nabholz, B.; Bonneau, M.; Carrio, E.M.; Nadachowska-Brzyska, K.; Ellegren, H.; Galtier, N. Life History Traits, Protein Evolution, and the Nearly Neutral Theory in Amniotes. Mol. Biol. Evol. 2016, 33, 1517–1527. [Google Scholar] [CrossRef]
- Schield, D.R.; Card, D.C.; Hales, N.R.; Perry, B.W.; Pasquesi, G.M.; Blackmon, H.; Adams, R.H.; Corbin, A.B.; Smith, C.F.; Ramesh, B.; et al. The Origins and Evolution of Chromosomes, Dosage Compensation, and Mechanisms Underlying Venom Regulation in Snakes. Genome Res. 2019, 29, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Cree, A.; Thompson, M.B.; Daugherty, C.H. Tuatara Sex Determination. Nature 1995, 375, 543. [Google Scholar] [CrossRef]
- Mitchell, N.J.; Nelson, N.J.; Cree, A.; Pledger, S.; Keall, S.N.; Daugherty, C.H. Support for a Rare Pattern of Temperature-Dependent Sex Determination in Archaic Reptiles: Evidence from Two Species of Tuatara (Sphenodon). Front. Zool. 2006, 3, 9. [Google Scholar] [CrossRef]
- González, E.J.; Martínez-López, M.; Morales-Garduza, M.A.; García-Morales, R.; Charruau, P.; Gallardo-Cruz, J.A. The Sex-Determination Pattern in Crocodilians: A Systematic Review of Three Decades of Research. J. Anim. Ecol. 2019, 88, 1417–1427. [Google Scholar] [CrossRef]
- Ewert, M.A.; Nelson, C.E. Sex Determination in Turtles: Diverse Patterns and Some Possible Adaptive Values. Copeia 1991, 1991, 50–69. [Google Scholar] [CrossRef]
- Viets, B.E.; Ewert, M.A.; Talent, L.G.; Nelson, C.E. Sex-Determining Mechanisms in Squamate Reptiles. J. Exp. Zool. 1994, 270, 45–56. [Google Scholar] [CrossRef]
- Pokorná, M.; Kratochvíl, L. Phylogeny of Sex-Determining Mechanisms in Squamate Reptiles: Are Sex Chromosomes an Evolutionary Trap? Zool. J. Linn. Soc. 2009, 156, 168–183. [Google Scholar] [CrossRef]
- Bista, B.; Valenzuela, N. Turtle Insights into the Evolution of the Reptilian Karyotype and the Genomic Architecture of Sex Determination. Genes 2020, 11, 416. [Google Scholar] [CrossRef] [PubMed]
- Robert, K.A.; Thompson, M.B. Viviparous Lizard Selects Sex of Embryos. Nature 2001, 412, 698–699. [Google Scholar] [CrossRef]
- Shine, R.; Elphick, M.J.; Donnellan, S. Co-Occurrence of Multiple, Supposedly Incompatible Modes of Sex Determination in a Lizard Population. Ecol. Lett. 2002, 5, 486–489. [Google Scholar] [CrossRef]
- Quinn, A.E.; Georges, A.; Sarre, S.D.; Guarino, F.; Ezaz, T.; Graves, J.A.M. Temperature Sex Reversal Implies Sex Gene Dosage in a Reptile. Science 2007, 316, 411. [Google Scholar] [CrossRef] [PubMed]
- Radder, R.S.; Quinn, A.E.; Georges, A.; Sarre, S.D.; Shine, R. Genetic Evidence for Co-Occurrence of Chromosomal and Thermal Sex-Determining Systems in a Lizard. Biol. Lett. 2008, 4, 176–178. [Google Scholar] [CrossRef]
- Holleley, C.E.; O’Meally, D.; Sarre, S.D.; Marshall Graves, J.A.; Ezaz, T.; Matsubara, K.; Azad, B.; Zhang, X.; Georges, A. Sex Reversal Triggers the Rapid Transition from Genetic to Temperature-Dependent Sex. Nature 2015, 523, 79–82. [Google Scholar] [CrossRef]
- Hill, P.L.; Burridge, C.P.; Ezaz, T.; Wapstra, E. Conservation of Sex-Linked Markers among Conspecific Populations of a Viviparous Skink, Niveoscincus ocellatus, Exhibiting Genetic and Temperature-Dependent Sex Determination. Genome Biol. Evol. 2018, 10, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Páramo, P.; Dissanayake, D.S.B.; Lira-Noriega, A.; Martínez-Pacheco, M.L.; Acosta, A.; Ramírez-Suástegui, C.; Méndez-de-la-Cruz, F.R.; Székely, T.; Urrutia, A.O.; Georges, A.; et al. Viviparous Reptile Regarded to Have Temperature-Dependent Sex Determination Has Old XY Chromosomes. Genome Biol. Evol. 2020, 12, 924–930. [Google Scholar] [CrossRef]
- Blackburn, D.G. Classification of the Reproductive Patterns of Amniotes. Herpetol. Monogr. 2000, 14, 371–377. [Google Scholar] [CrossRef]
- Sites, J.W.; Reeder, T.W.; Wiens, J.J. Phylogenetic Insights on Evolutionary Novelties in Lizards and Snakes: Sex, Birth, Bodies, Niches, and Venom. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 227–244. [Google Scholar] [CrossRef]
- Whittington, C.M.; Van Dyke, J.U.; Liang, S.Q.T.; Edwards, S.V.; Shine, R.; Thompson, M.B.; Grueber, C.E. Understanding the Evolution of Viviparity Using Intraspecific Variation in Reproductive Mode and Transitional Forms of Pregnancy. Biol. Rev. 2022, 97, 1179–1192. [Google Scholar] [CrossRef]
- Pyron, R.A.; Burbrink, F.T. Early Origin of Viviparity and Multiple Reversions to Oviparity in Squamate Reptiles. Ecol. Lett. 2014, 17, 13–21. [Google Scholar] [CrossRef]
- Brana, F.; Bea, A. Biomodalité de La Reproduction Chez Lacerta vivipara (Reptilia, Lacertidae). Bull. Société Herpétologique Fr. 1987, 44, 1–5. [Google Scholar]
- Heulin, B. Données Nouvelles Sur Les Populations Ovipares de Lacerta vivipara. C. R. Académie Sci. Sér. 3 Sci. Vie 1988, 306, 63–68. [Google Scholar]
- Qualls, C.P.; Shine, R.; Donnellan, S.; Hutchinson, M. The Evolution of Viviparity within the Australian Scincid Lizard Lerista bougainvillii. J. Zool. 1995, 237, 13–26. [Google Scholar] [CrossRef]
- Smith, S.A.; Shine, R. Intraspecific Variation in Reproductive Mode within the Scincid Lizard Saiphos equalis. Aust. J. Zool. 1997, 45, 435–445. [Google Scholar] [CrossRef]
- Braz, H.B.; Scartozzoni, R.R.; Almeida-Santos, S.M. Reproductive Modes of the South American Water Snakes: A Study System for the Evolution of Viviparity in Squamate Reptiles. Zool. Anz. J. Comp. Zool. 2016, 263, 33–44. [Google Scholar] [CrossRef]
- Recknagel, H.; Carruthers, M.; Yurchenko, A.A.; Nokhbatolfoghahai, M.; Kamenos, N.A.; Bain, M.M.; Elmer, K.R. The Functional Genetic Architecture of Egg-Laying and Live-Bearing Reproduction in Common Lizards. Nat. Ecol. Evol. 2021, 5, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Shine, R. A New Hypothesis for the Evolution of Viviparity in Reptiles. Am. Nat. 1995, 145, 809–823. [Google Scholar] [CrossRef]
- Noble, D.W.A.; Stenhouse, V.; Schwanz, L.E. Developmental Temperatures and Phenotypic Plasticity in Reptiles: A Systematic Review and Meta-Analysis. Biol. Rev. 2018, 93, 72–97. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, O.; Kluge, A.G. Natural Parthenogenesis in the Gekkonid Lizard Lepidodactylus lugubris. J. Genet. 1972, 61, 14–26. [Google Scholar] [CrossRef]
- Kluge, A.G.; Eckardt, M.J. Hemidactylus garnotii Duméril and Bibron, a Triploid All-Female Species of Gekkonid Lizard. Copeia 1969, 1969, 651–664. [Google Scholar] [CrossRef]
- McDowell, S.B. A Catalogue of the Snakes of New Guinea and the Solomons, with Special Reference to Those in the Bernice P. Bishop Museum, Part I. Scolecophidia. J. Herpetol. 1974, 8, 1–57. [Google Scholar] [CrossRef]
- Lowe, C.H.; Wright, J.W. Evolution of Parthenogenetic Species of Cnemidophorus (Whiptail Lizards) in Western North America. J. Ariz. Acad. Sci. 1966, 4, 81–87. [Google Scholar]
- Darevsky, I. Natural Parthenogenesis in Certain Subspecies of Rock Lizard, Lacerta saxicola. Dokl. Akad. Nauk SSSR 1958, 122, 730–732. [Google Scholar]
- Watts, P.C.; Buley, K.R.; Sanderson, S.; Boardman, W.; Ciofi, C.; Gibson, R. Parthenogenesis in Komodo Dragons. Nature 2006, 444, 1021–1022. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, W.E. Production of an Embryo by an Acrochordus javanicus Isolated for Seven Years. Copeia 1979, 1979, 744–745. [Google Scholar] [CrossRef]
- Schuett, G.W.; Fernandez, P.J.; Gergits, W.F.; Casna, N.J.; Chiszar, D.; Smith, H.M.; Mitton, J.B.; Mackessy, S.P.; Odum, R.A.; Demlong, M.J. Production of Offspring in the Absence of Males: Evidence for Facultative Parthenogenesis in Bisexual Snakes. Herpetol. Nat. Hist. 1997, 5, 1–10. [Google Scholar]
- Groot, T.V.M.; Bruins, E.; Breeuwer, J.A.J. Molecular Genetic Evidence for Parthenogenesis in the Burmese Python, Python molurus bivittatus. Heredity 2003, 90, 130–135. [Google Scholar] [CrossRef]
- Booth, W.; Johnson, D.H.; Moore, S.; Schal, C.; Vargo, E.L. Evidence for Viable, Non-Clonal but Fatherless Boa Constrictors. Biol. Lett. 2011, 7, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Booth, W.; Million, L.; Reynolds, R.G.; Burghardt, G.M.; Vargo, E.L.; Schal, C.; Tzika, A.C.; Schuett, G.W. Consecutive Virgin Births in the New World Boid Snake, the Colombian Rainbow Boa, Epicrates maurus. J. Hered. 2011, 102, 759–763. [Google Scholar] [CrossRef][Green Version]
- Reynolds, R.G.; Booth, W.; Schuett, G.W.; Fitzpatrick, B.M.; Burghardt, G.M. Successive Virgin Births of Viable Male Progeny in the Checkered Gartersnake, Thamnophis marcianus. Biol. J. Linn. Soc. 2012, 107, 566–572. [Google Scholar] [CrossRef]
- Card, D.C.; Vonk, F.J.; Smalbrugge, S.; Casewell, N.R.; Wüster, W.; Castoe, T.A.; Schuett, G.W.; Booth, W. Genome-Wide Data Implicate Terminal Fusion Automixis in King Cobra Facultative Parthenogenesis. Sci. Rep. 2021, 11, 7271. [Google Scholar] [CrossRef]
- Booth, W.; Smith, C.F.; Eskridge, P.H.; Hoss, S.K.; Mendelson, J.R.; Schuett, G.W. Facultative Parthenogenesis Discovered in Wild Vertebrates. Biol. Lett. 2012, 8, 983–985. [Google Scholar] [CrossRef]
- Gregory, T.R. Coincidence, Coevolution, or Causation? DNA Content, Cell size, and the C-Value Enigma. Biol. Rev. 2001, 76, 65–101. [Google Scholar] [CrossRef] [PubMed]
- Waltari, E.; Edwards, S.V. Evolutionary Dynamics of Intron Size, Genome Size, and Physiological Correlates in Archosaurs. Am. Nat. 2002, 160, 539–552. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gregory, T.R. Genome Size Evolution in Animals. In The Evolution of the Genome; Gregory, T.R., Ed.; Academic Press: Burlington, VT, USA, 2005; pp. 3–87. ISBN 978-0-12-301463-4. [Google Scholar]
- Organ, C.L.; Moreno, R.G.; Edwards, S.V. Three Tiers of Genome Evolution in Reptiles. Integr. Comp. Biol. 2008, 48, 494–504. [Google Scholar] [CrossRef]
- Oliver, M.J.; Petrov, D.; Ackerly, D.; Falkowski, P.; Schofield, O.M. The Mode and Tempo of Genome Size Evolution in Eukaryotes. Genome Res. 2007, 17, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Olmo, E. Rate of Chromosome Changes and Speciation in Reptiles. Genetica 2005, 125, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, A. Micro versus Macro: A Review of Structure and Functions of Avian Micro-and Macrochromosomes. Russ. J. Genet. 1996, 32, 517–527. [Google Scholar]
- Hillier, L.W.; Miller, W.; Birney, E.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.A.M.; Delany, M.E.; et al. Sequence and Comparative Analysis of the Chicken Genome Provide Unique Perspectives on Vertebrate Evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef]
- Smith, J.; Bruley, C.K.; Paton, I.R.; Dunn, I.; Jones, C.T.; Windsor, D.; Morrice, D.R.; Law, A.S.; Masabanda, J.; Sazanov, A.; et al. Differences in Gene Density on Chicken Macrochromosomes and Microchromosomes. Anim. Genet. 2000, 31, 96–103. [Google Scholar] [CrossRef]
- Rodionov, A.V.; Chel’sheva, L.A.; Soloveĭ, I.V.; Miakoshina, I.A. Chiasma distribution in the lampbrush chromosomes of the chicken Gallus gallus domesticus: Hot spots of recombination and their possible role in proper dysjunction of homologous chromosomes at the first meiotic division. Genetika 1992, 28, 151–160. [Google Scholar] [PubMed]
- Perry, B.W.; Schield, D.R.; Adams, R.H.; Castoe, T.A. Microchromosomes Exhibit Distinct Features of Vertebrate Chromosome Structure and Function with Underappreciated Ramifications for Genome Evolution. Mol. Biol. Evol. 2021, 38, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Waters, P.D.; Patel, H.R.; Ruiz-Herrera, A.; Álvarez-González, L.; Lister, N.C.; Simakov, O.; Ezaz, T.; Kaur, P.; Frere, C.; Grützner, F.; et al. Microchromosomes Are Building Blocks of Bird, Reptile, and Mammal Chromosomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2112494118. [Google Scholar] [CrossRef]
- Fredga, K. Chromosomal Changes in Vertebrate Evolution. Proc. R. Soc. Lond. B Biol. Sci. 1977, 199, 377–397. [Google Scholar]
- Damas, J.; Corbo, M.; Lewin, H.A. Vertebrate Chromosome Evolution. Annu. Rev. Anim. Biosci. 2021, 9, 1–27. [Google Scholar] [CrossRef]
- Srikulnath, K.; Ahmad, S.F.; Singchat, W.; Panthum, T. Why Do Some Vertebrates Have Microchromosomes? Cells 2021, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Sex-Chromosome Evolution: Recent Progress and the Influence of Male and Female Heterogamety. Nat. Rev. Genet. 2011, 12, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Dean, R.; Zimmer, F.; Mank, J.E. How to Make a Sex Chromosome. Nat. Commun. 2016, 7, 12087. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.K.; Nordén, A.K.; Hansson, B. Sex Chromosome Evolution: Historical Insights and Future Perspectives. Proc. R. Soc. B Biol. Sci. 2017, 284, 20162806. [Google Scholar] [CrossRef] [PubMed]
- Van Doorn, G.S.; Kirkpatrick, M. Turnover of Sex Chromosomes Induced by Sexual Conflict. Nature 2007, 449, 909–912. [Google Scholar] [CrossRef]
- Wright, A.E.; Darolti, I.; Bloch, N.I.; Oostra, V.; Sandkam, B.; Buechel, S.D.; Kolm, N.; Breden, F.; Vicoso, B.; Mank, J.E. Convergent Recombination Suppression Suggests Role of Sexual Selection in Guppy Sex Chromosome Formation. Nat. Commun. 2017, 8, 14251. [Google Scholar] [CrossRef] [PubMed]
- Vicoso, B. Molecular and Evolutionary Dynamics of Animal Sex-Chromosome Turnover. Nat. Ecol. Evol. 2019, 3, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.; Coryell, J.; Ezaz, T.; Lynch, J.; Scantlebury, D.P.; Zarkower, D. Restriction Site-Associated DNA Sequencing (RAD-Seq) Reveals an Extraordinary Number of Transitions among Gecko Sex-Determining Systems. Mol. Biol. Evol. 2015, 32, 1296–1309. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.; Castoe, T.A.; Nielsen, S.V.; Banks, J.L.; Card, D.C.; Schield, D.R.; Schuett, G.W.; Booth, W. The Discovery of XY Sex Chromosomes in a Boa and Python. Curr. Biol. 2017, 27, 2148–2153. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Sex Chromosomes and Sex-Linked Genes; Monographs on Endocrinology; Springer: Berlin/Heidelberg, Germany, 1967; ISBN 978-3-642-88178-7. [Google Scholar]
- Matsubara, K.; Tarui, H.; Toriba, M.; Yamada, K.; Nishida-Umehara, C.; Agata, K.; Matsuda, Y. Evidence for Different Origin of Sex Chromosomes in Snakes, Birds, and Mammals and Step-Wise Differentiation of Snake Sex Chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 18190–18195. [Google Scholar] [CrossRef]
- Castoe, T.A.; de Koning, A.P.J.; Kim, H.-M.; Gu, W.; Noonan, B.P.; Naylor, G.; Jiang, Z.J.; Parkinson, C.L.; Pollock, D.D. Evidence for an Ancient Adaptive Episode of Convergent Molecular Evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8986–8991. [Google Scholar] [CrossRef]
- Macey, J.R.; Pabinger, S.; Barbieri, C.G.; Buring, E.S.; Gonzalez, V.L.; Mulcahy, D.G.; DeMeo, D.P.; Urban, L.; Hime, P.M.; Prost, S.; et al. Evidence of Two Deeply Divergent Co-Existing Mitochondrial Genomes in the Tuatara Reveals an Extremely Complex Genomic Organization. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef]
- Jiang, Z.J.; Castoe, T.A.; Austin, C.C.; Burbrink, F.T.; Herron, M.D.; McGuire, J.A.; Parkinson, C.L.; Pollock, D.D. Comparative Mitochondrial Genomics of Snakes: Extraordinary Substitution Rate Dynamics and Functionality of the Duplicate Control Region. BMC Evol. Biol. 2007, 7, 123. [Google Scholar] [CrossRef]
- Castoe, T.A.; Jiang, Z.J.; Gu, W.; Wang, Z.O.; Pollock, D.D. Adaptive Evolution and Functional Redesign of Core Metabolic Proteins in Snakes. PLoS ONE 2008, 3, e2201. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Chinwalla, A.T.; Cook, L.L.; Delehaunty, K.D.; Fewell, G.A.; Fulton, L.A.; Fulton, R.S.; Graves, T.A.; Hillier, L.W.; Mardis, E.R.; McPherson, J.D.; et al. Initial Sequencing and Comparative Analysis of the Mouse Genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.J.; Fairley, S.; et al. The Genome of a Songbird. Nature 2010, 464, 757–762. [Google Scholar] [CrossRef]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative Genomics Reveals Insights into Avian Genome Evolution and Adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Suh, A. Evolution of Bird Genomes—A Transposon’s-Eye View. Ann. N. Y. Acad. Sci. 2017, 1389, 164–185. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, A.M. Phylogenomic Investigation of CR1 LINE Diversity in Reptiles. Syst. Biol. 2006, 55, 902–911. [Google Scholar] [CrossRef]
- Shedlock, A.M.; Botka, C.W.; Zhao, S.; Shetty, J.; Zhang, T.; Liu, J.S.; Deschavanne, P.J.; Edwards, S.V. Phylogenomics of Nonavian Reptiles and the Structure of the Ancestral Amniote Genome. Proc. Natl. Acad. Sci. USA 2007, 104, 2767–2772. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; Hall, K.T.; Guibotsy Mboulas, M.L.; Gu, W.; de Koning, A.P.J.; Fox, S.E.; Poole, A.W.; Vemulapalli, V.; Daza, J.M.; Mockler, T.; et al. Discovery of Highly Divergent Repeat Landscapes in Snake Genomes Using High-Throughput Sequencing. Genome Biol. Evol. 2011, 3, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; de Koning, A.P.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese Python Genome Reveals the Molecular Basis for Extreme Adaptation in Snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef]
- Di-Poï, N.; Montoya-Burgos, J.I.; Miller, H.; Pourquié, O.; Milinkovitch, M.C.; Duboule, D. Changes in Hox Genes’ Structure and Function during the Evolution of the Squamate Body Plan. Nature 2010, 464, 99–103. [Google Scholar] [CrossRef]
- Shaney, K.J.; Card, D.C.; Schield, D.R.; Ruggiero, R.P.; Pollock, D.D.; Mackessy, S.P.; Castoe, T.A. Squamate Reptile Genomics and Evolution. In Toxinology: Venom Genomics and Proteomics; Gopalakrishnakone, P., Calvete, J.J., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2014; pp. 1–18. ISBN 978-94-007-6649-5. [Google Scholar]
- Perry, B.W.; Gopalan, S.S.; Pasquesi, G.I.M.; Schield, D.R.; Westfall, A.K.; Smith, C.F.; Koludarov, I.; Chippindale, P.T.; Pellegrino, M.W.; Chuong, E.B.; et al. Snake Venom Gene Expression Is Coordinated by Novel Regulatory Architecture and the Integration of Multiple Co-Opted Vertebrate Pathways. Genome Res. 2022, 32, 1058–1073. [Google Scholar] [CrossRef]
- Kordiš, D.; Gubenšek, F. Bov-B Long Interspersed Repeated DNA (LINE) Sequences Are Present in Vipera ammodytes Phospholipase A2 Genes and in Genomes of Viperidae Snakes. Eur. J. Biochem. 1997, 246, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Kordiš, D.; Gubenšek, F. Unusual Horizontal Transfer of a Long Interspersed Nuclear Element between Distant Vertebrate Classes. Proc. Natl. Acad. Sci. USA 1998, 95, 10704–10709. [Google Scholar] [CrossRef]
- Kordiš, D.; Gubenšek, F. Molecular Evolution of Bov-B LINEs in Vertebrates. Gene 1999, 238, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Piskurek, O.; Okada, N. Poxviruses as Possible Vectors for Horizontal Transfer of Retroposons from Reptiles to Mammals. Proc. Natl. Acad. Sci. USA 2007, 104, 12046–12051. [Google Scholar] [CrossRef]
- Pace, J.K.; Gilbert, C.; Clark, M.S.; Feschotte, C. Repeated Horizontal Transfer of a DNA Transposon in Mammals and Other Tetrapods. Proc. Natl. Acad. Sci. USA 2008, 105, 17023–17028. [Google Scholar] [CrossRef]
- Novick, P.A.; Basta, H.; Floumanhaft, M.; McClure, M.A.; Boissinot, S. The Evolutionary Dynamics of Autonomous Non-LTR Retrotransposons in the Lizard Anolis carolinensis Shows More Similarity to Fish Than Mammals. Mol. Biol. Evol. 2009, 26, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Piskurek, O.; Nishihara, H.; Okada, N. The Evolution of Two Partner LINE/SINE Families and a Full-Length Chromodomain-Containing Ty3/Gypsy LTR Element in the First Reptilian Genome of Anolis carolinensis. Gene 2009, 441, 111–118. [Google Scholar] [CrossRef]
- Novick, P.; Smith, J.; Ray, D.; Boissinot, S. Independent and Parallel Lateral Transfer of DNA Transposons in Tetrapod Genomes. Gene 2010, 449, 85–94. [Google Scholar] [CrossRef]
- Gilbert, C.; Hernandez, S.S.; Flores-Benabib, J.; Smith, E.N.; Feschotte, C. Rampant Horizontal Transfer of SPIN Transposons in Squamate Reptiles. Mol. Biol. Evol. 2012, 29, 503–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Walsh, A.M.; Kortschak, R.D.; Gardner, M.G.; Bertozzi, T.; Adelson, D.L. Widespread Horizontal Transfer of Retrotransposons. Proc. Natl. Acad. Sci. USA 2013, 110, 1012–1016. [Google Scholar] [CrossRef]
- Bernardi, G. The Vertebrate Genome: Isochores and Evolution. Mol. Biol. Evol. 1993, 10, 186–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernardi, G. Isochores and the Evolutionary Genomics of Vertebrates. Gene 2000, 241, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, S.M.; Bernardo Carvalho, A.; Clark, A.G. Local Rates of Recombination Are Positively Correlated with GC Content in the Human Genome. Mol. Biol. Evol. 2001, 18, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, D.; D’Onofrio, G.; Aïssani, B.; Macaya, G.; Gautier, C.; Bernardi, G. The Distribution of Genes in the Human Genome. Gene 1991, 100, 181–187. [Google Scholar] [CrossRef]
- Jabbari, K.; Bernardi, G. CpG Doublets, CpG Islands and Alu Repeats in Long Human DNA Sequences from Different Isochore Families. Gene 1998, 224, 123–128. [Google Scholar] [CrossRef]
- Duret, L.; Mouchiroud, D.; Gautier, C. Statistical Analysis of Vertebrate Sequences Reveals That Long Genes Are Scarce in GC-Rich Isochores. J. Mol. Evol. 1995, 40, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Fujiyama, A.; Ichiba, Y.; Hattori, M.; Yada, T.; Sakaki, Y.; Ikemura, T. Chromosome-Wide Assessment of Replication Timing for Human Chromosomes 11q and 21q: Disease-Related Genes in Timing-Switch Regions. Hum. Mol. Genet. 2002, 11, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Piganeau, G.; Mouchiroud, D.; Duret, L. GC-Content Evolution in Mammalian Genomes: The Biased Gene Conversion Hypothesis. Genetics 2001, 159, 907–911. [Google Scholar] [CrossRef]
- Galtier, N. Gene Conversion Drives GC Content Evolution in Mammalian Histones. Trends Genet. 2003, 19, 65–68. [Google Scholar] [CrossRef]
- Marais, G. Biased Gene Conversion: Implications for Genome and Sex Evolution. Trends Genet. 2003, 19, 330–338. [Google Scholar] [CrossRef]
- Meunier, J.; Duret, L. Recombination Drives the Evolution of GC-Content in the Human Genome. Mol. Biol. Evol. 2004, 21, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Galtier, N.; Duret, L.; Glémin, S.; Ranwez, V. GC-Biased Gene Conversion Promotes the Fixation of Deleterious Amino Acid Changes in Primates. Trends Genet. 2009, 25, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Schield, D.R.; Pasquesi, G.I.M.; Perry, B.W.; Adams, R.H.; Nikolakis, Z.L.; Westfall, A.K.; Orton, R.W.; Meik, J.M.; Mackessy, S.P.; Castoe, T.A. Snake Recombination Landscapes Are Concentrated in Functional Regions despite PRDM9. Mol. Biol. Evol. 2020, 37, 1272–1294. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Clay, O.; Bernardi, G. Compositional Patterns in Reptilian Genomes. Gene 2002, 295, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Zelus, D.; Mouchiroud, D. Warm-Blooded Isochore Structure in Nile Crocodile and Turtle. Mol. Biol. Evol. 1999, 16, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Fortes, G.G.; Bouza, C.; Martínez, P.; Sánchez, L. Diversity in Isochore Structure among Cold-Blooded Vertebrates Based on GC Content of Coding and Non-Coding Sequences. Genetica 2007, 129, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Landan, G.; Graur, D. Can GC Content at Third-Codon Positions Be Used as a Proxy for Isochore Composition? Mol. Biol. Evol. 2009, 26, 1829–1833. [Google Scholar] [CrossRef]
- Zhang, C.-T.; Zhang, R. Isochore Structures in the Mouse Genome. Genomics 2004, 83, 384–394. [Google Scholar] [CrossRef]
- Alföldi, J.; Di Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D.; et al. The Genome of the Green Anole Lizard and a Comparative Analysis with Birds and Mammals. Nature 2011, 477, 587–591. [Google Scholar] [CrossRef]
- Bravo, G.A.; Schmitt, C.J.; Edwards, S.V. What Have We Learned from the First 500 Avian Genomes? Annu. Rev. Ecol. Evol. Syst. 2021, 52, 611–639. [Google Scholar] [CrossRef]
- Moritz, C.C.; Pratt, R.C.; Bank, S.; Bourke, G.; Bragg, J.G.; Doughty, P.; Keogh, J.S.; Laver, R.J.; Potter, S.; Teasdale, L.C.; et al. Cryptic Lineage Diversity, Body Size Divergence, and Sympatry in a Species Complex of Australian Lizards (Gehyra). Evolution 2018, 72, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Oliver, P.M.; Clegg, J.R.; Fisher, R.N.; Richards, S.J.; Taylor, P.N.; Jocque, M.M.T. A New Biogeographically Disjunct Giant Gecko (Gehyra: Gekkonidae: Reptilia) from the East Melanesian Islands. Zootaxa 2016, 4208, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Doughty, P.; Bourke, G.; Tedeschi, L.G.; Pratt, R.C.; Oliver, P.M.; Palmer, R.A.; Moritz, C. Species Delimitation in the Gehyra nana (Squamata: Gekkonidae) Complex: Cryptic and Divergent Morphological Evolution in the Australian Monsoonal Tropics, with the Description of Four New Species. Zootaxa 2018, 4403, 201. [Google Scholar] [CrossRef]
- Oliver, P.M.; Prasetya, A.M.; Tedeschi, L.G.; Fenker, J.; Ellis, R.J.; Doughty, P.; Moritz, C. Crypsis and Convergence: Integrative Taxonomic Revision of the Gehyra australis Group (Squamata: Gekkonidae) from Northern Australia. PeerJ 2020, 8, e7971. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Douglas, M.R.; Schuett, G.W.; Beck, D.D.; Sullivan, B.K. Conservation Phylogenetics of Helodermatid Lizards Using Multiple Molecular Markers and a Supertree Approach. Mol. Phylogenet. Evol. 2010, 55, 153–167. [Google Scholar] [CrossRef]
- Hugall, A.F.; Foster, R.; Hutchinson, M.; Lee, M.S.Y. Phylogeny of Australasian Agamid Lizards Based on Nuclear and Mitochondrial Genes: Implications for Morphological Evolution and Biogeography. Biol. J. Linn. Soc. 2008, 93, 343–358. [Google Scholar] [CrossRef]
- Edwards, T.; Tollis, M.; Hsieh, P.; Gutenkunst, R.N.; Liu, Z.; Kusumi, K.; Culver, M.; Murphy, R.W. Assessing Models of Speciation under Different Biogeographic Scenarios; an Empirical Study Using Multi-Locus and RNA-Seq Analyses. Ecol. Evol. 2016, 6, 379–396. [Google Scholar] [CrossRef]
- Edwards, T.; Karl, A.E.; Vaughn, M.; Rosen, P.C.; Torres, C.M.; Murphy, R.W. The Desert Tortoise Trichotomy: Mexico Hosts a Third, New Sister-Species of Tortoise in the Gopherus morafkai–G. agassizii Group. ZooKeys 2016, 562, 131–158. [Google Scholar] [CrossRef]
- Spinks, P.Q.; Thomson, R.C.; McCartney-Melstad, E.; Shaffer, H.B. Phylogeny and Temporal Diversification of the New World Pond Turtles (Emydidae). Mol. Phylogenet. Evol. 2016, 103, 85–97. [Google Scholar] [CrossRef]
- Spinks, P.Q.; Thomson, R.C.; Zhang, Y.; Che, J.; Wu, Y.; Bradley Shaffer, H. Species Boundaries and Phylogenetic Relationships in the Critically Endangered Asian Box Turtle Genus Cuora. Mol. Phylogenet. Evol. 2012, 63, 656–667. [Google Scholar] [CrossRef]
- Zhou, H.; Jiang, Y.; Nie, L.; Yin, H.; Li, H.; Dong, X.; Zhao, F.; Zhang, H.; Pu, Y.; Huang, Z.; et al. The Historical Speciation of Mauremys Sensu Lato: Ancestral Area Reconstruction and Interspecific Gene Flow Level Assessment Provide New Insights. PLoS ONE 2015, 10, e0144711. [Google Scholar] [CrossRef]
- Figueroa, A.; McKelvy, A.D.; Grismer, L.L.; Bell, C.D.; Lailvaux, S.P. A Species-Level Phylogeny of Extant Snakes with Description of a New Colubrid Subfamily and Genus. PLoS ONE 2016, 11, e0161070. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Khaing, K.P.P.; Than, N.L.; Baranski, D.; Schell, T.; Greve, C.; Janke, A.; Pauls, S.U. A New Genus and Species of Mud Snake from Myanmar (Reptilia, Squamata, Homalopsidae). Zootaxa 2021, 4915, 301–325. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Losos, J. Lizards in an Evolutionary Tree: Ecology and Adaptive Radiation of Anoles; University of California Press: Berkeley, CA, USA, 2011; ISBN 978-0-520-26984-2. [Google Scholar]
- Stuart, Y.E.; Campbell, T.S.; Hohenlohe, P.A.; Reynolds, R.G.; Revell, L.J.; Losos, J.B. Rapid Evolution of a Native Species Following Invasion by a Congener. Science 2014, 346, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Staton, S.C.; Cheviron, Z.A.; Rochette, N.; Catchen, J.; Losos, J.B.; Edwards, S.V. Winter Storms Drive Rapid Phenotypic, Regulatory, and Genomic Shifts in the Green Anole Lizard. Science 2017, 357, 495–498. [Google Scholar] [CrossRef]
- Card, D.C.; Van Camp, A.G.; Santonastaso, T.; Jensen-Seaman, M.I.; Anthony, N.M.; Edwards, S.V. Structure and Evolution of the Squamate Major Histocompatibility Complex as Revealed by Two Anolis Lizard Genomes. Front. Genet. 2022, 13, 979746. [Google Scholar] [CrossRef]
- Geneva, A.J.; Park, S.; Bock, D.G.; de Mello, P.L.H.; Sarigol, F.; Tollis, M.; Donihue, C.M.; Reynolds, R.G.; Feiner, N.; Rasys, A.M.; et al. Chromosome-Scale Genome Assembly of the Brown Anole (Anolis sagrei), an Emerging Model Species. Commun. Biol. 2022, 5, 1–13. [Google Scholar] [CrossRef]
- Kanamori, S.; Díaz, L.M.; Cádiz, A.; Yamaguchi, K.; Shigenobu, S.; Kawata, M. Draft Genome of Six Cuban Anolis Lizards and Insights into Genetic Changes during Their Diversification. BMC Ecol. Evol. 2022, 22, 129. [Google Scholar] [CrossRef]
- Couzin, J. NSF’s Ark Draws Alligators, Algae, and Wasps. Science 2002, 297, 1638–1639. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Miyake, T.; Edwards, S.V.; Amemiya, C.T. Tuatara (Sphenodon) Genomics: BAC Library Construction, Sequence Survey, and Application to the DMRT Gene Family. J. Hered. 2006, 97, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Janes, D.E.; Organ, C.; Valenzuela, N. New Resources Inform Study of Genome Size, Content, and Organization in Nonavian Reptiles. Integr. Comp. Biol. 2008, 48, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Badenhorst, D.; Hillier, L.W.; Literman, R.; Montiel, E.E.; Radhakrishnan, S.; Shen, Y.; Minx, P.; Janes, D.E.; Warren, W.C.; Edwards, S.V.; et al. Physical Mapping and Refinement of the Painted Turtle Genome (Chrysemys picta) Inform Amniote Genome Evolution and Challenge Turtle-Bird Chromosomal Conservation. Genome Biol. Evol. 2015, 7, 2038–2050. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomson, R.C.; Shedlock, A.M.; Edwards, S.V.; Shaffer, H.B. Developing Markers for Multilocus Phylogenetics in Non-Model Organisms: A Test Case with Turtles. Mol. Phylogenet. Evol. 2008, 49, 514–525. [Google Scholar] [CrossRef]
- Shedlock, A.M.; Janes, D.E.; Edwards, S.V. Amniote Phylogenomics: Testing Evolutionary Hypotheses with BAC Library Scanning and Targeted Clone Analysis of Large-Scale DNA Sequences from Reptiles. In Phylogenomics; Murphy, W.J., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2008; pp. 91–117. ISBN 978-1-59745-581-7. [Google Scholar]
- Janes, D.E.; Chapus, C.; Gondo, Y.; Clayton, D.F.; Sinha, S.; Blatti, C.A.; Organ, C.L.; Fujita, M.K.; Balakrishnan, C.N.; Edwards, S.V. Reptiles and Mammals Have Differentially Retained Long Conserved Noncoding Sequences from the Amniote Ancestor. Genome Biol. Evol. 2011, 3, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Shendure, J.; Mitra, R.D.; Varma, C.; Church, G.M. Advanced Sequencing Technologies: Methods and Goals. Nat. Rev. Genet. 2004, 5, 335–344. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-Generation DNA Sequencing Methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate Whole Human Genome Sequencing Using Reversible Terminator Chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Cronn, R.; Liston, A.; Parks, M.; Gernandt, D.S.; Shen, R.; Mockler, T. Multiplex Sequencing of Plant Chloroplast Genomes Using Solexa Sequencing-by-Synthesis Technology. Nucleic Acids Res. 2008, 36, e122. [Google Scholar] [CrossRef] [PubMed]
- Degnan, J.H.; Rosenberg, N.A. Gene Tree Discordance, Phylogenetic Inference and the Multispecies Coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.V.; Beerli, P. Perspective: Gene Divergence, Population Divergence, and the Variance in Coalescence Time in Phylogeographic Studies. Evolution 2000, 54, 1839–1854. [Google Scholar] [CrossRef]
- Arbogast, B.S.; Edwards, S.V.; Wakeley, J.; Beerli, P.; Slowinski, J.B. Estimating Divergence Times from Molecular Data on Phylogenetic and Population Genetic Timescales. Annu. Rev. Ecol. Syst. 2002, 33, 707–740. [Google Scholar] [CrossRef]
- Wakeley, J. Coalescent Theory: An Introduction; W.H. Freeman: New York, NY, USA, 2009; ISBN 978-0-9747077-5-4. [Google Scholar]
- Wakeley, J.; King, L.; Low, B.S.; Ramachandran, S. Gene Genealogies within a Fixed Pedigree, and the Robustness of Kingman’s Coalescent. Genetics 2012, 190, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Pluzhnikov, A.; Donnelly, P. Optimal Sequencing Strategies for Surveying Molecular Genetic Diversity. Genetics 1996, 144, 1247–1262. [Google Scholar] [CrossRef]
- Felsenstein, J. Accuracy of Coalescent Likelihood Estimates: Do We Need More Sites, More Sequences, or More Loci? Mol. Biol. Evol. 2006, 23, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Jennings, W.B. On the Independent Gene Trees Assumption in Phylogenomic Studies. Mol. Ecol. 2017, 26, 4862–4871. [Google Scholar] [CrossRef]
- Jennings, W.B.; Edwards, S.V. Speciational History of Australian Grass Finches (Poephila) Inferred from Thirty Gene Trees. Evolution 2005, 59, 2033–2047. [Google Scholar] [CrossRef]
- Lee, J.Y.; Edwards, S.V. Divergence Across Australia’s Carpentarian Barrier: Statistical Phylogeography of the Red-Backed Fairy Wren (Malurus melanocephalus). Evolution 2008, 62, 3117–3134. [Google Scholar] [CrossRef]
- Smith, B.T.; Harvey, M.G.; Faircloth, B.C.; Glenn, T.C.; Brumfield, R.T. Target Capture and Massively Parallel Sequencing of Ultraconserved Elements for Comparative Studies at Shallow Evolutionary Time Scales. Syst. Biol. 2014, 63, 83–95. [Google Scholar] [CrossRef]
- Costa, I.R.; Prosdocimi, F.; Jennings, W.B. In Silico Phylogenomics Using Complete Genomes: A Case Study on the Evolution of Hominoids. Genome Res. 2016, 26, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Good, J.M. Reduced Representation Methods for Subgenomic Enrichment and Next-Generation Sequencing. In Molecular Methods for Evolutionary Genetics; Orgogozo, V., Rockman, M.V., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 85–103. ISBN 978-1-61779-228-1. [Google Scholar]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution Hybrid Selection with Ultra-Long Oligonucleotides for Massively Parallel Targeted Sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef]
- Andermann, T.; Torres Jiménez, M.F.; Matos-Maraví, P.; Batista, R.; Blanco-Pastor, J.L.; Gustafsson, A.L.S.; Kistler, L.; Liberal, I.M.; Oxelman, B.; Bacon, C.D.; et al. A Guide to Carrying Out a Phylogenomic Target Sequence Capture Project. Front. Genet. 2020, 10, 1047. [Google Scholar] [CrossRef]
- Faircloth, B.C.; McCormack, J.E.; Crawford, N.G.; Harvey, M.G.; Brumfield, R.T.; Glenn, T.C. Ultraconserved Elements Anchor Thousands of Genetic Markers Spanning Multiple Evolutionary Timescales. Syst. Biol. 2012, 61, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, A.R.; Emme, S.A.; Lemmon, E.M. Anchored Hybrid Enrichment for Massively High-Throughput Phylogenomics. Syst. Biol. 2012, 61, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Crawford, N.G.; Faircloth, B.C.; McCormack, J.E.; Brumfield, R.T.; Winker, K.; Glenn, T.C. More than 1000 Ultraconserved Elements Provide Evidence That Turtles Are the Sister Group of Archosaurs. Biol. Lett. 2012, 8, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Brandley, M.C.; Bragg, J.G.; Singhal, S.; Chapple, D.G.; Jennings, C.K.; Lemmon, A.R.; Lemmon, E.M.; Thompson, M.B.; Moritz, C. Evaluating the Performance of Anchored Hybrid Enrichment at the Tips of the Tree of Life: A Phylogenetic Analysis of Australian Eugongylus Group Scincid Lizards. BMC Evol. Biol. 2015, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Leaché, A.D.; Chavez, A.S.; Jones, L.N.; Grummer, J.A.; Gottscho, A.D.; Linkem, C.W. Phylogenomics of Phrynosomatid Lizards: Conflicting Signals from Sequence Capture versus Restriction Site Associated DNA Sequencing. Genome Biol. Evol. 2015, 7, 706–719. [Google Scholar] [CrossRef]
- Bejerano, G.; Pheasant, M.; Makunin, I.; Stephen, S.; Kent, W.J.; Mattick, J.S.; Haussler, D. Ultraconserved Elements in the Human Genome. Science 2004, 304, 1321–1325. [Google Scholar] [CrossRef]
- Singhal, S.; Grundler, M.; Colli, G.; Rabosky, D.L. Squamate Conserved Loci (SqCL): A Unified Set of Conserved Loci for Phylogenomics and Population Genetics of Squamate Reptiles. Mol. Ecol. Resour. 2017, 17, e12–e24. [Google Scholar] [CrossRef] [PubMed]
- Wiens, J.J.; Hutter, C.R.; Mulcahy, D.G.; Noonan, B.P.; Townsend, T.M.; Sites, J.W.; Reeder, T.W. Resolving the Phylogeny of Lizards and Snakes (Squamata) with Extensive Sampling of Genes and Species. Biol. Lett. 2012, 8, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A Phylogeny and Revised Classification of Squamata, Including 4161 Species of Lizards and Snakes. BMC Evol. Biol. 2013, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.D.; Thomas, W.K.; Meyer, A.; Edwards, S.V.; Pääbo, S.; Villablanca, F.X.; Wilson, A.C. Dynamics of Mitochondrial DNA Evolution in Animals: Amplification and Sequencing with Conserved Primers. Proc. Natl. Acad. Sci. USA 1989, 86, 6196–6200. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M.; Moritz, C.; Mable, B.K. Molecular Systematics, 2nd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 1996; ISBN 978-0-87893-282-5. [Google Scholar]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000; ISBN 978-0-674-66638-2. [Google Scholar]
- Ashman, L.G.; Bragg, J.G.; Doughty, P.; Hutchinson, M.N.; Bank, S.; Matzke, N.J.; Oliver, P.; Moritz, C. Diversification across Biomes in a Continental Lizard Radiation. Evolution 2018, 72, 1553–1569. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.; Bryson, R.W., Jr.; García-Vázquez, U.O.; Nieto-Montes De Oca, A.; Lazcano, D.; Mccormack, J.E.; Klicka, J. Phylogenomics of Alligator Lizards Elucidate Diversification Patterns across the Mexican Transition Zone and Support the Recognition of a New Genus. Biol. J. Linn. Soc. 2022, 135, 25–39. [Google Scholar] [CrossRef]
- Blom, M.P.K.; Bragg, J.G.; Potter, S.; Moritz, C. Accounting for Uncertainty in Gene Tree Estimation: Summary-Coalescent Species Tree Inference in a Challenging Radiation of Australian Lizards. Syst. Biol. 2017, 66, 352–366. [Google Scholar] [CrossRef]
- Blom, M.P.K.; Matzke, N.J.; Bragg, J.G.; Arida, E.; Austin, C.C.; Backlin, A.R.; Carretero, M.A.; Fisher, R.N.; Glaw, F.; Hathaway, S.A.; et al. Habitat Preference Modulates Trans-Oceanic Dispersal in a Terrestrial Vertebrate. Proc. Royal Soc. B 2019, 286, 20182575. [Google Scholar] [CrossRef]
- Bragg, J.G.; Potter, S.; Afonso Silva, A.C.; Hoskin, C.J.; Bai, B.Y.H.; Moritz, C. Phylogenomics of a Rapid Radiation: The Australian Rainbow Skinks. BMC Evol. Biol. 2018, 18, 15. [Google Scholar] [CrossRef]
- Brennan, I.G.; Lemmon, A.R.; Lemmon, E.M.; Portik, D.M.; Weijola, V.; Welton, L.; Donnellan, S.C.; Keogh, J.S. Phylogenomics of Monitor Lizards and the Role of Competition in Dictating Body Size Disparity. Syst. Biol. 2021, 70, 120–132. [Google Scholar] [CrossRef]
- Bryson, R.W., Jr.; Linkem, C.W.; Pavón-Vázquez, C.J.; Nieto-Montes de Oca, A.; Klicka, J.; McCormack, J.E. A Phylogenomic Perspective on the Biogeography of Skinks in the Plestiodon brevirostris Group Inferred from Target Enrichment of Ultraconserved Elements. J. Biogeogr. 2017, 44, 2033–2044. [Google Scholar] [CrossRef]
- Domingos, F.M.C.B.; Colli, G.R.; Lemmon, A.; Lemmon, E.M.; Beheregaray, L.B. In the Shadows: Phylogenomics and Coalescent Species Delimitation Unveil Cryptic Diversity in a Cerrado Endemic Lizard (Squamata: Tropidurus). Mol. Phylogenet. Evol. 2017, 107, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Freitas, S.; Westram, A.M.; Schwander, T.; Arakelyan, M.; Ilgaz, Ç.; Kumlutas, Y.; Harris, D.J.; Carretero, M.A.; Butlin, R.K. Parthenogenesis in Darevskia Lizards: A Rare Outcome of Common Hybridization, not a Common Outcome of Rare Hybridization. Evolution 2022, 76, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Porta, J.; Irisarri, I.; Kirchner, M.; Rodríguez, A.; Kirchhof, S.; Brown, J.L.; MacLeod, A.; Turner, A.P.; Ahmadzadeh, F.; Albaladejo, G.; et al. Environmental Temperatures Shape Thermal Physiology as Well as Diversification and Genome-Wide Substitution Rates in Lizards. Nat. Commun. 2019, 10, 4077. [Google Scholar] [CrossRef]
- Grummer, J.A.; Morando, M.M.; Avila, L.J.; Sites, J.W.; Leaché, A.D. Phylogenomic Evidence for a Recent and Rapid Radiation of Lizards in the Patagonian Liolaemus fitzingerii Species Group. Mol. Phylogenet. Evol. 2018, 125, 243–254. [Google Scholar] [CrossRef]
- Morando, M.; Olave, M.; Avila, L.J.; Sites, J.W.; Leaché, A.D. Phylogenomic Data Resolve Higher-Level Relationships within South American Liolaemus Lizards. Mol. Phylogenet. Evol. 2020, 147, 106781. [Google Scholar] [CrossRef]
- Panzera, A.; Leaché, A.D.; D’Elía, G.; Victoriano, P.F. Phylogenomic Analysis of the Chilean Clade of Liolaemus Lizards (Squamata: Liolaemidae) Based on Sequence Capture Data. PeerJ 2017, 5, e3941. [Google Scholar] [CrossRef]
- Ramírez-Reyes, T.; Blair, C.; Flores-Villela, O.; Piñero, D.; Lathrop, A.; Murphy, R. Phylogenomics and Molecular Species Delimitation Reveals Great Cryptic Diversity of Leaf-Toed Geckos (Phyllodactylidae: Phyllodactylus), Ancient Origins, and Diversification in Mexico. Mol. Phylogenet. Evol. 2020, 150, 106880. [Google Scholar] [CrossRef]
- Reilly, S.B.; Stubbs, A.L.; Arida, E.; Karin, B.R.; Arifin, U.; Kaiser, H.; Bi, K.; Iskandar, D.T.; McGuire, J.A. Phylogenomic Analysis Reveals Dispersal-Driven Speciation and Divergence with Gene Flow in Lesser Sunda Flying Lizards (Genus Draco). Syst. Biol. 2022, 71, 221–241. [Google Scholar] [CrossRef]
- Reilly, S.B.; Karin, B.R.; Stubbs, A.L.; Arida, E.; Arifin, U.; Kaiser, H.; Bi, K.; Hamidy, A.; Iskandar, D.T.; McGuire, J.A. Diverge and Conquer: Phylogenomics of Southern Wallacean Forest Skinks (Genus: Sphenomorphus) and Their Colonization of the Lesser Sunda Archipelago. Evolution 2022, 76, 2281–2301. [Google Scholar] [CrossRef]
- Reynolds, R.G.; Miller, A.H.; Pasachnik, S.A.; Knapp, C.R.; Welch, M.E.; Colosimo, G.; Gerber, G.P.; Drawert, B.; Iverson, J.B. Phylogenomics and Historical Biogeography of West Indian Rock Iguanas (Genus Cyclura). Mol. Phylogenet. Evol. 2022, 174, 107548. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Z.B.; Perkins, S.L.; Austin, C.C. Multiple Origins of Green Blood in New Guinea Lizards. Sci. Adv. 2018, 4, eaao5017. [Google Scholar] [CrossRef] [PubMed]
- Schools, M.; Kasprowicz, A.; Blair Hedges, S. Phylogenomic Data Resolve the Historical Biogeography and Ecomorphs of Neotropical Forest Lizards (Squamata, Diploglossidae). Mol. Phylogenet. Evol. 2022, 175, 107577. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Hoskin, C.J.; Couper, P.; Potter, S.; Moritz, C. A Framework for Resolving Cryptic Species: A Case Study from the Lizards of the Australian Wet Tropics. Syst. Biol. 2018, 67, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Skipwith, P.L.; Bi, K.; Oliver, P.M. Relicts and Radiations: Phylogenomics of an Australasian Lizard Clade with East Gondwanan Origins (Gekkota: Diplodactyloidea). Mol. Phylogenet. Evol. 2019, 140, 106589. [Google Scholar] [CrossRef] [PubMed]
- Tucker, D.B.; Hedges, S.B.; Colli, G.R.; Pyron, R.A.; Sites, J.W., Jr. Genomic Timetree and Historical Biogeography of Caribbean Island Ameiva Lizards (Pholidoscelis: Teiidae). Ecol. Evol. 2017, 7, 7080–7090. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Guo, X.; Travers, S.L.; Su, Y.-C.; Olson, K.V.; Bauer, A.M.; Grismer, L.L.; Siler, C.D.; Moyle, R.G.; Andersen, M.J.; et al. Parachute Geckos Free Fall into Synonymy: Gekko Phylogeny, and a New Subgeneric Classification, Inferred from Thousands of Ultraconserved Elements. Mol. Phylogenet. Evol. 2020, 146, 106731. [Google Scholar] [CrossRef]
- Zozaya, S.M.; Teasdale, L.C.; Tedeschi, L.G.; Higgie, M.; Hoskin, C.J.; Moritz, C. Initiation of Speciation across Multiple Dimensions in a Rock-Restricted, Tropical Lizard. Mol. Ecol. 2022, 32, 680–695. [Google Scholar] [CrossRef]
- Bernstein, J.M.; Ruane, S. Maximizing Molecular Data from Low-Quality Fluid-Preserved Specimens in Natural History Collections. Front. Ecol. Evol. 2022, 10, 581. [Google Scholar] [CrossRef]
- Blair, C.; Bryson, R.W., Jr.; Linkem, C.W.; Lazcano, D.; Klicka, J.; McCormack, J.E. Cryptic Diversity in the Mexican Highlands: Thousands of UCE Loci Help Illuminate Phylogenetic Relationships, Species Limits and Divergence Times of Montane Rattlesnakes (Viperidae: Crotalus). Mol. Ecol. Resour. 2019, 19, 349–365. [Google Scholar] [CrossRef]
- Chen, X.; Lemmon, A.R.; Lemmon, E.M.; Pyron, R.A.; Burbrink, F.T. Using Phylogenomics to Understand the Link between Biogeographic Origins and Regional Diversification in Ratsnakes. Mol. Phylogenet. Evol. 2017, 111, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Esquerré, D.; Donnellan, S.; Brennan, I.G.; Lemmon, A.R.; Moriarty Lemmon, E.; Zaher, H.; Grazziotin, F.G.; Keogh, J.S. Phylogenomics, Biogeography, and Morphometrics Reveal Rapid Phenotypic Evolution in Pythons after Crossing Wallace’s Line. Syst. Biol. 2020, 69, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Hallas, J.M.; Parchman, T.L.; Feldman, C.R. Phylogenomic Analyses Resolve Relationships among Garter Snakes (Thamnophis: Natricinae: Colubridae) and Elucidate Biogeographic History and Morphological Evolution. Mol. Phylogenet. Evol. 2022, 167, 107374. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, D.; Zhang, P. Simultaneously Collecting Coding and Non-Coding Phylogenomic Data Using Homemade Full-Length CDNA Probes, Tested by Resolving the High-Level Relationships of Colubridae. Front. Ecol. Evol. 2022, 10, 969581. [Google Scholar] [CrossRef]
- Myers, E.A.; Mulcahy, D.G.; Falk, B.; Johnson, K.; Carbi, M.; de Queiroz, K. Interspecific Gene Flow and Mitochondrial Genome Capture during the Radiation of Jamaican Anolis Lizards (Squamata; Iguanidae). Syst. Biol. 2022, 71, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Natusch, D.J.D.; Esquerré, D.; Lyons, J.A.; Hamidy, A.; Lemmon, A.R.; Lemmon, E.M.; Riyanto, A.; Keogh, J.S.; Donnellan, S. Phylogenomics, Biogeography and Taxonomic Revision of New Guinean Pythons (Pythonidae, Leiopython) Harvested for International Trade. Mol. Phylogenet. Evol. 2021, 158, 106960. [Google Scholar] [CrossRef] [PubMed]
- Nikolakis, Z.L.; Orton, R.W.; Crother, B.I. Fine-Scale Population Structure within an Eastern Nearctic Snake Complex (Pituophis melanoleucus). Zool. Scr. 2022, 51, 133–146. [Google Scholar] [CrossRef]
- Ruane, S.; Austin, C.C. Phylogenomics Using Formalin-Fixed and 100+ Year-Old Intractable Natural History Specimens. Mol. Ecol. Resour. 2017, 17, 1003–1008. [Google Scholar] [CrossRef]
- Burbrink, F.T.; Grazziotin, F.G.; Pyron, R.A.; Cundall, D.; Donnellan, S.; Irish, F.; Keogh, J.S.; Kraus, F.; Murphy, R.W.; Noonan, B.; et al. Interrogating Genomic-Scale Data for Squamata (Lizards, Snakes, and Amphisbaenians) Shows no Support for Key Traditional Morphological Relationships. Syst. Biol. 2020, 69, 502–520. [Google Scholar] [CrossRef]
- Singhal, S.; Colston, T.J.; Grundler, M.R.; Smith, S.A.; Costa, G.C.; Colli, G.R.; Moritz, C.; Pyron, R.A.; Rabosky, D.L. Congruence and Conflict in the Higher-Level Phylogenetics of Squamate Reptiles: An Expanded Phylogenomic Perspective. Syst. Biol. 2021, 70, 542–557. [Google Scholar] [CrossRef]
- Streicher, J.W.; Wiens, J.J. Phylogenomic Analyses of More than 4000 Nuclear Loci Resolve the Origin of Snakes among Lizard Families. Biol. Lett. 2017, 13, 20170393. [Google Scholar] [CrossRef]
- Shaffer, H.B.; McCartney-Melstad, E.; Near, T.J.; Mount, G.G.; Spinks, P.Q. Phylogenomic Analyses of 539 Highly Informative Loci Dates a Fully Resolved Time Tree for the Major Clades of Living Turtles (Testudines). Mol. Phylogenet. Evol. 2017, 115, 7–15. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Davey, J.W.; Blaxter, M.L. RADSeq: Next-Generation Population Genetics. Brief. Funct. Genomics 2010, 9, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Etter, P.D.; Bassham, S.; Hohenlohe, P.A.; Johnson, E.A.; Cresko, W.A. SNP Discovery and Genotyping for Evolutionary Genetics Using RAD Sequencing. In Molecular Methods for Evolutionary Genetics; Orgogozo, V., Rockman, M.V., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; pp. 157–178. ISBN 978-1-61779-228-1. [Google Scholar]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef]
- Arnold, B.; Corbett-Detig, R.B.; Hartl, D.; Bomblies, K. RADseq Underestimates Diversity and Introduces Genealogical Biases Due to Nonrandom Haplotype Sampling. Mol. Ecol. 2013, 22, 3179–3190. [Google Scholar] [CrossRef]
- Cariou, M.; Duret, L.; Charlat, S. Is RAD-seq Suitable for Phylogenetic Inference? An In Silico Assessment and Optimization. Ecol. Evol. 2013, 3, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.W.; Hejnol, A.; Matus, D.Q.; Pang, K.; Browne, W.E.; Smith, S.A.; Seaver, E.; Rouse, G.W.; Obst, M.; Edgecombe, G.D.; et al. Broad Phylogenomic Sampling Improves Resolution of the Animal Tree of Life. Nature 2008, 452, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; Vanderpool, D.; Singhal, S.; Linderoth, T.; Moritz, C.; Good, J.M. Transcriptome-Based Exon Capture Enables Highly Cost-Effective Comparative Genomic Data Collection at Moderate Evolutionary Scales. BMC Genom. 2012, 13, 403. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; Linderoth, T.; Vanderpool, D.; Good, J.M.; Nielsen, R.; Moritz, C. Unlocking the Vault: Next-Generation Museum Population Genomics. Mol. Ecol. 2013, 22, 6018–6032. [Google Scholar] [CrossRef]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-Genome Analyses Resolve Early Branches in the Tree of Life of Modern Birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, H.B.; Toffelmier, E.; Corbett-Detig, R.B.; Escalona, M.; Erickson, B.; Fiedler, P.; Gold, M.; Harrigan, R.J.; Hodges, S.; Luckau, T.K.; et al. Landscape Genomics to Enable Conservation Actions: The California Conservation Genomics Project. J. Hered. 2022, 113, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Grismer, J.L.; Escalona, M.; Miller, C.; Beraut, E.; Fairbairn, C.W.; Marimuthu, M.P.A.; Nguyen, O.; Toffelmier, E.; Wang, I.J.; Shaffer, H.B. Reference Genome of the Rubber Boa, Charina bottae (Serpentes: Boidae). J. Hered. 2022, 113, 641–648. [Google Scholar] [CrossRef]
- Todd, B.D.; Jenkinson, T.S.; Escalona, M.; Beraut, E.; Nguyen, O.; Sahasrabudhe, R.; Scott, P.A.; Toffelmier, E.; Wang, I.J.; Shaffer, H.B. Reference Genome of the Northwestern Pond Turtle, Actinemys marmorata. J. Hered. 2022, 113, 624–631. [Google Scholar] [CrossRef]
- Wood, D.A.; Richmond, J.Q.; Escalona, M.; Marimuthu, M.P.A.; Nguyen, O.; Sacco, S.; Beraut, E.; Westphal, M.; Fisher, R.N.; Vandergast, A.G.; et al. Reference Genome of the California Glossy Snake, Arizona elegans occidentalis: A Declining California Species of Special Concern. J. Hered. 2022, 113, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Bravo, G.A.; Antonelli, A.; Bacon, C.D.; Bartoszek, K.; Blom, M.P.K.; Huynh, S.; Jones, G.; Knowles, L.L.; Lamichhaney, S.; Marcussen, T.; et al. Embracing Heterogeneity: Coalescing the Tree of Life and the Future of Phylogenomics. PeerJ 2019, 7, e6399. [Google Scholar] [CrossRef]
- McCormack, J.E.; Faircloth, B.C.; Crawford, N.G.; Gowaty, P.A.; Brumfield, R.T.; Glenn, T.C. Ultraconserved Elements Are Novel Phylogenomic Markers That Resolve Placental Mammal Phylogeny When Combined with Species-Tree Analysis. Genome Res. 2012, 22, 746–754. [Google Scholar] [CrossRef]
- Karl, S.A.; Avise, J.C. PCR-Based Assays of Mendelian Polymorphisms from Anonymous Single-Copy Nuclear DNA: Techniques and Applications for Population Genetics. Mol. Biol. Evol. 1993, 10, 342–361. [Google Scholar] [CrossRef][Green Version]
- Thomson, R.C.; Wang, I.J.; Johnson, J.R. Genome-Enabled Development of DNA Markers for Ecology, Evolution and Conservation. Mol. Ecol. 2010, 19, 2184–2195. [Google Scholar] [CrossRef]
- Reilly, S.B.; Marks, S.B.; Jennings, W.B. Defining Evolutionary Boundaries across Parapatric Ecomorphs of Black Salamanders (Aneides flavipunctatus) with Conservation Implications. Mol. Ecol. 2012, 21, 5745–5761. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Smith, N.J.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef]
- Scheet, P.; Stephens, M. A Fast and Flexible Statistical Model for Large-Scale Population Genotype Data: Applications to Inferring Missing Genotypes and Haplotypic Phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Andermann, T.; Fernandes, A.M.; Olsson, U.; Töpel, M.; Pfeil, B.; Oxelman, B.; Aleixo, A.; Faircloth, B.C.; Antonelli, A. Allele Phasing Greatly Improves the Phylogenetic Utility of Ultraconserved Elements. Syst. Biol. 2019, 68, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Bennett, J.; Flouri, T.; Leaché, A.D.; Yang, Z. Phase Resolution of Heterozygous Sites in Diploid Genomes Is Important to Phylogenomic Analysis under the Multispecies Coalescent Model. Syst. Biol. 2022, 71, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Card, D.C.; Shapiro, B.; Giribet, G.; Moritz, C.; Edwards, S.V. Museum Genomics. Annu. Rev. Genet. 2021, 55, 633–659. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.V.; Cloutier, A.; Baker, A.J. Conserved Nonexonic Elements: A Novel Class of Marker for Phylogenomics. Syst. Biol. 2017, 66, 1028–1044. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Edwards, S.V.; Liu, L. The Multispecies Coalescent Model Outperforms Concatenation Across Diverse Phylogenomic Data Sets. Syst. Biol. 2020, 69, 795–812. [Google Scholar] [CrossRef]
- Edwards, S.V. Is a New and General Theory of Molecular Systematics Emerging? Evol. Int. J. Org. Evol. 2009, 63, 1–19. [Google Scholar] [CrossRef]
- Brito, P.H.; Edwards, S.V. Multilocus Phylogeography and Phylogenetics Using Sequence-Based Markers. Genetica 2009, 135, 439–455. [Google Scholar] [CrossRef]
- Beerli, P.; Mashayekhi, S.; Sadeghi, M.; Khodaei, M.; Shaw, K. Population Genetic Inference with MIGRATE. Curr. Protoc. Bioinforma. 2019, 68, e87. [Google Scholar] [CrossRef]
- Flouri, T.; Jiao, X.; Rannala, B.; Yang, Z. A Bayesian Implementation of the Multispecies Coalescent Model with Introgression for Phylogenomic Analysis. Mol. Biol. Evol. 2020, 37, 1211–1223. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Kimball, R.T.; Braun, E.L. Introns Outperform Exons in Analyses of Basal Avian Phylogeny Using Clathrin Heavy Chain Genes. Gene 2008, 410, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.-L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why Do Phylogenomic Data Sets Yield Conflicting Trees? Data Type Influences the Avian Tree of Life More than Taxon Sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.; Hickey, G.; Diekhans, M.; Fiddes, I.T.; Novak, A.M.; Deran, A.; Fang, Q.; Xie, D.; Feng, S.; Stiller, J.; et al. Progressive Cactus Is a Multiple-Genome Aligner for the Thousand-Genome Era. Nature 2020, 587, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Guarracino, A.; Heumos, S.; Nahnsen, S.; Prins, P.; Garrison, E. ODGI: Understanding Pangenome Graphs. Bioinformatics 2022, 38, 3319–3326. [Google Scholar] [CrossRef]
- Garrison, E.; Guarracino, A. Unbiased Pangenome Graphs. Bioinformatics 2022, 39, btac743. [Google Scholar] [CrossRef]
- Smith, S.D.; Pennell, M.W.; Dunn, C.W.; Edwards, S.V. Phylogenetics Is the New Genetics (for Most of Biodiversity). Trends Ecol. Evol. 2020, 35, 415–425. [Google Scholar] [CrossRef]
- Pease, J.B.; Haak, D.C.; Hahn, M.W.; Moyle, L.C. Phylogenomics Reveals Three Sources of Adaptive Variation during a Rapid Radiation. PLoS Biol. 2016, 14, e1002379. [Google Scholar] [CrossRef]
- McLean, C.Y.; Reno, P.L.; Pollen, A.A.; Bassan, A.I.; Capellini, T.D.; Guenther, C.; Indjeian, V.B.; Lim, X.H.; Menke, D.B.; Schaar, B.T.; et al. Human-Specific Loss of Regulatory DNA and the Evolution of Human-Specific Traits. Nature 2011, 471, 216–219. [Google Scholar] [CrossRef]
- Hiller, M.; Schaar, B.T.; Indjeian, V.B.; Kingsley, D.M.; Hagey, L.R.; Bejerano, G. A “Forward Genomics” Approach Links Genotype to Phenotype Using Independent Phenotypic Losses among Related Species. Cell. Rep. 2012, 2, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Marcovitz, A.; Jia, R.; Bejerano, G. “Reverse Genomics” Predicts Function of Human Conserved Noncoding Elements. Mol. Biol. Evol. 2016, 33, 1358–1369. [Google Scholar] [CrossRef] [PubMed]
- Marcovitz, A.; Turakhia, Y.; Gloudemans, M.; Braun, B.A.; Chen, H.I.; Bejerano, G. A Novel Unbiased Test for Molecular Convergent Evolution and Discoveries in Echolocating, Aquatic and High-Altitude Mammals. bioRxiv 2017, 1, 170985. [Google Scholar] [CrossRef]
- Sackton, T.B.; Grayson, P.; Cloutier, A.; Hu, Z.; Liu, J.S.; Wheeler, N.E.; Gardner, P.P.; Clarke, J.A.; Baker, A.J.; Clamp, M.; et al. Convergent Regulatory Evolution and Loss of Flight in Paleognathous Birds. Science 2019, 364, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Muntané, G.; Farré, X.; Rodríguez, J.A.; Pegueroles, C.; Hughes, D.A.; de Magalhães, J.P.; Gabaldón, T.; Navarro, A. Biological Processes Modulating Longevity across Primates: A Phylogenetic Genome-Phenome Analysis. Mol. Biol. Evol. 2018, 35, 1990–2004. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Partha, R.; Clark, N.L.; Chikina, M. Pan-Mammalian Analysis of Molecular Constraints Underlying Extended Lifespan. eLife 2020, 9, e51089. [Google Scholar] [CrossRef]
- Kolora, S.R.R.; Owens, G.L.; Vazquez, J.M.; Stubbs, A.; Chatla, K.; Jainese, C.; Seeto, K.; McCrea, M.; Sandel, M.W.; Vianna, J.A.; et al. Origins and Evolution of Extreme Life Span in Pacific Ocean Rockfishes. Science 2021, 374, 842–847. [Google Scholar] [CrossRef]
- Treaster, S.; Deelen, J.; Daane, J.M.; Murabito, J.; Karasik, D.; Harris, M.P. Convergent Genomics of Longevity in Rockfishes Highlights the Genetics of Human Life Span Variation. Sci. Adv. 2023, 9, eadd2743. [Google Scholar] [CrossRef]
- Roscito, J.G.; Sameith, K.; Kirilenko, B.M.; Hecker, N.; Winkler, S.; Dahl, A.; Rodrigues, M.T.; Hiller, M. Convergent and Lineage-Specific Genomic Differences in Limb Regulatory Elements in Limbless Reptile Lineages. Cell Rep. 2022, 38, 110280. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Poujol, R. A Phylogenetic Model for Investigating Correlated Evolution of Substitution Rates and Continuous Phenotypic Characters. Mol. Biol. Evol. 2011, 28, 729–744. [Google Scholar] [CrossRef]
- Hu, Z.; Sackton, T.B.; Edwards, S.V.; Liu, J.S. Bayesian Detection of Convergent Rate Changes of Conserved Noncoding Elements on Phylogenetic Trees. Mol. Biol. Evol. 2019, 36, 1086–1100. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Meyer, W.K.; Partha, R.; Mao, W.; Clark, N.L.; Chikina, M. RERconverge: An R Package for Associating Evolutionary Rates with Convergent Traits. Bioinformatics 2019, 35, 4815–4817. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Hu, Z.; Thomas, G.; Edwards, S.V.; Sackton, T.B.; Liu, J.S. PhyloAcc-GT: A Bayesian Method for Inferring Patterns of Substitution Rate Shifts and Associations with Binary Traits under Gene Tree Discordance. bioRxiv 2022, 1, 2022.12.23.521765. [Google Scholar] [CrossRef]
- Shine, R. Life-History Evolution in Reptiles. Annu. Rev. Ecol. Evol. Syst. 2005, 36, 23–46. [Google Scholar] [CrossRef]
- Brandley, M.C.; Huelsenbeck, J.P.; Wiens, J.J. Rates and Patterns in the Evolution of Snake-Like Body Form in Squamate Reptiles: Evidence for Repeated Re-Evolution of Lost Digits and Long-Term Persistence of Intermediate Body Forms. Evolution 2008, 62, 2042–2064. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Taxon | Type of Loci | # of Loci | # Samples | Depth of Divergences |
|---|---|---|---|---|---|
| Ashman et al., 2018 [209] | lizards | exons | 547 | 64 | shallow |
| Blair et al., 2022 [210] | lizards | UCEs | 3157 | 34 | shallow |
| Blom et al., 2017 [211] | lizards | exons | 2840 | 28 | shallow |
| Blom et al., 2019 [212] | lizards | exons | 2457 | 135 | shallow |
| Bragg et al., 2018 [213] | lizards | exons | 2364 | 123 | shallow-deep |
| Brennan et al., 2021 [214] | lizards | AHEs | 388 | 103 | shallow-deep |
| Bryson et al., 2017 [215] | lizards | UCEs | 3282 | 58 | shallow |
| Domingos et al., 2017 [216] | lizards | AHEs | 422 | 30 | shallow |
| Freitas et al., 2022 [217] | lizards | exons | 625 | 69 | shallow |
| Garcia-Porta et al., 2019 [218] | lizards | AHEs + other | 6593 (324 AHEs + 6269 other) | 262 | shallow-deep |
| Grummer et al., 2018 [219] | lizards | UCEs + exons | 589 (541 UCEs + 44 exons) | 29 | shallow |
| Morando et al., 2020 [220] | lizards | UCEs + exons | 588 (540 UCEs + 44 exons) | 26 | deep |
| Moritz et al., 2018 [149] | lizards | exons | 1636 | 56 | shallow |
| Panzera et al., 2017 [221] | lizards | UCEs | 581 (538 UCEs + 43 exons) | 16 | higher |
| Ramírez-Reyes et al., 2020 [222] | lizards | RAD-seq | 78,970–549,193 | 90 | shallow |
| Reilly et al., 2022a [223] | lizards | exons | 709 | 99 | shallow-deep |
| Reilly et al., 2022b [224] | lizards | exons | 1154 | 104 | shallow |
| Reynolds et al., 2022 [225] | lizards | UCEs | 4055 | 82 | shallow-deep |
| Rodriguez et al., 2018 [226] | lizards | UCEs | 2690 | 119 | shallow-deep |
| Schools et al., 2022 [227] | lizards | UCEs | 5060 | 30 | higher |
| Singhal et al., 2018 [228] | lizards | exons | 2668 | 25 | shallow |
| Skipwith et al., 2019 [229] | lizards | UCEs | 4268 | 290 | shallow-deep |
| Tucker et al., 2017 [230] | lizards | AHEs | 316 | 16 | shallow |
| Wood et al., 2020 [231] | lizards | UCEs | 772–4715 | 42 | deep |
| Zozaya et al., 2022 [232] | lizards | exons | 1429 | 33 | shallow |
| Bernstein and Ruane 2022 [233] | snakes | AHEs + UCEs + other | 1–642 UCEs, 1–39 AHEs, 2–11 other | 156 | shallow-deep |
| Blair et al., 2019 [234] | snakes | UCEs | 3384 | 54 | shallow |
| Chen et al., 2017 [235] | snakes | AHEs | 304 | 88 | shallow |
| Esquerré et al., 2020 [236] | snakes | AHEs | 376 | 50 | deep |
| Hallas et al., 2022 [237] | snakes | RAD-seq | 22,289–48,867 | 49 | shallow |
| Li et al., 2022 [238] | snakes | exons + other | 3023 (1948 exons + 1948 other) | 24 | deep |
| Myers et al., 2022 [239] | lizards | RAD-seq | 2950 | 74 | shallow |
| Natusch et al., 2021 [240] | snakes | AHEs | 421 | 19 | shallow |
| Nikolakis et al., 2022 [241] | snakes | UCEs | 3383–4146 | 43 | shallow |
| Ruane and Austin 2017 [242] | snakes | UCEs | 2318 | 10 | deep |
| Burbrink et al., 2020 [243] | squamates | AHEs | 394 | 289 | deep |
| Singhal et al., 2021 [244] | squamates | AHEs + UCEs + other | 5462 (372 AHEs + 5052 UCEs + 38 other) | 92 | deep |
| Streicher and Wiens 2017 [245] | squamates | UCEs | 2738 | 24 | deep |
| Shaffer et al., 2017 [246] | turtles | UCEs + other | 539 | 24 | deep |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Card, D.C.; Jennings, W.B.; Edwards, S.V. Genome Evolution and the Future of Phylogenomics of Non-Avian Reptiles. Animals 2023, 13, 471. https://doi.org/10.3390/ani13030471
Card DC, Jennings WB, Edwards SV. Genome Evolution and the Future of Phylogenomics of Non-Avian Reptiles. Animals. 2023; 13(3):471. https://doi.org/10.3390/ani13030471
Chicago/Turabian StyleCard, Daren C., W. Bryan Jennings, and Scott V. Edwards. 2023. "Genome Evolution and the Future of Phylogenomics of Non-Avian Reptiles" Animals 13, no. 3: 471. https://doi.org/10.3390/ani13030471
APA StyleCard, D. C., Jennings, W. B., & Edwards, S. V. (2023). Genome Evolution and the Future of Phylogenomics of Non-Avian Reptiles. Animals, 13(3), 471. https://doi.org/10.3390/ani13030471