
Whole-Genome Resequencing Reveals Selection Signatures of Abigar Cattle for Local Adaptation
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Data
2.2. Quality Control, Read Mapping and Variant Calling
2.3. Genomic Diversity and Population Structure
2.4. Linkage Disequilibrium and Run of Homozygosity
2.5. Genome-Wide Selection Sweeps
3. Results
3.1. Sequence Reads and Variant Statistics
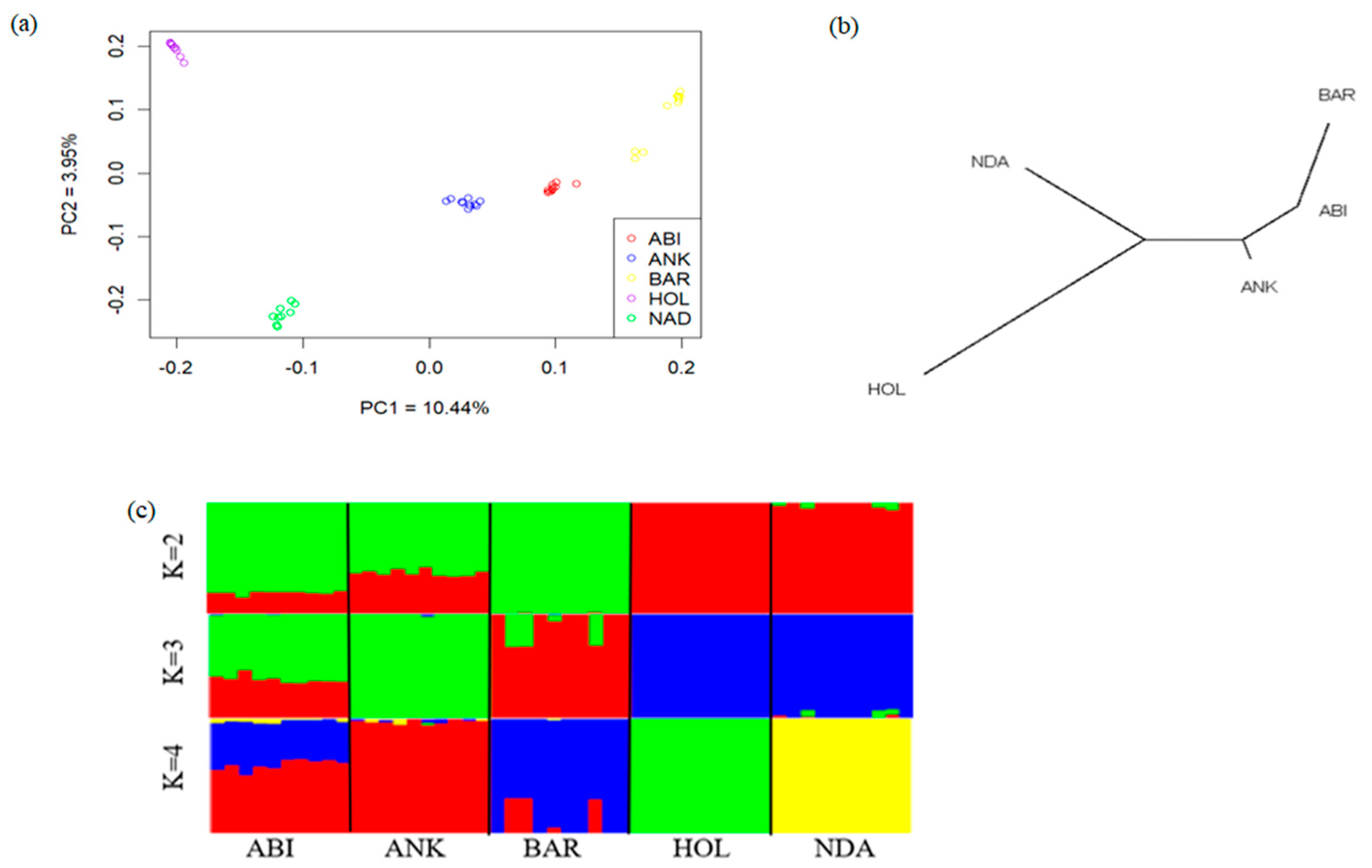
3.2. Population Genetic Structure
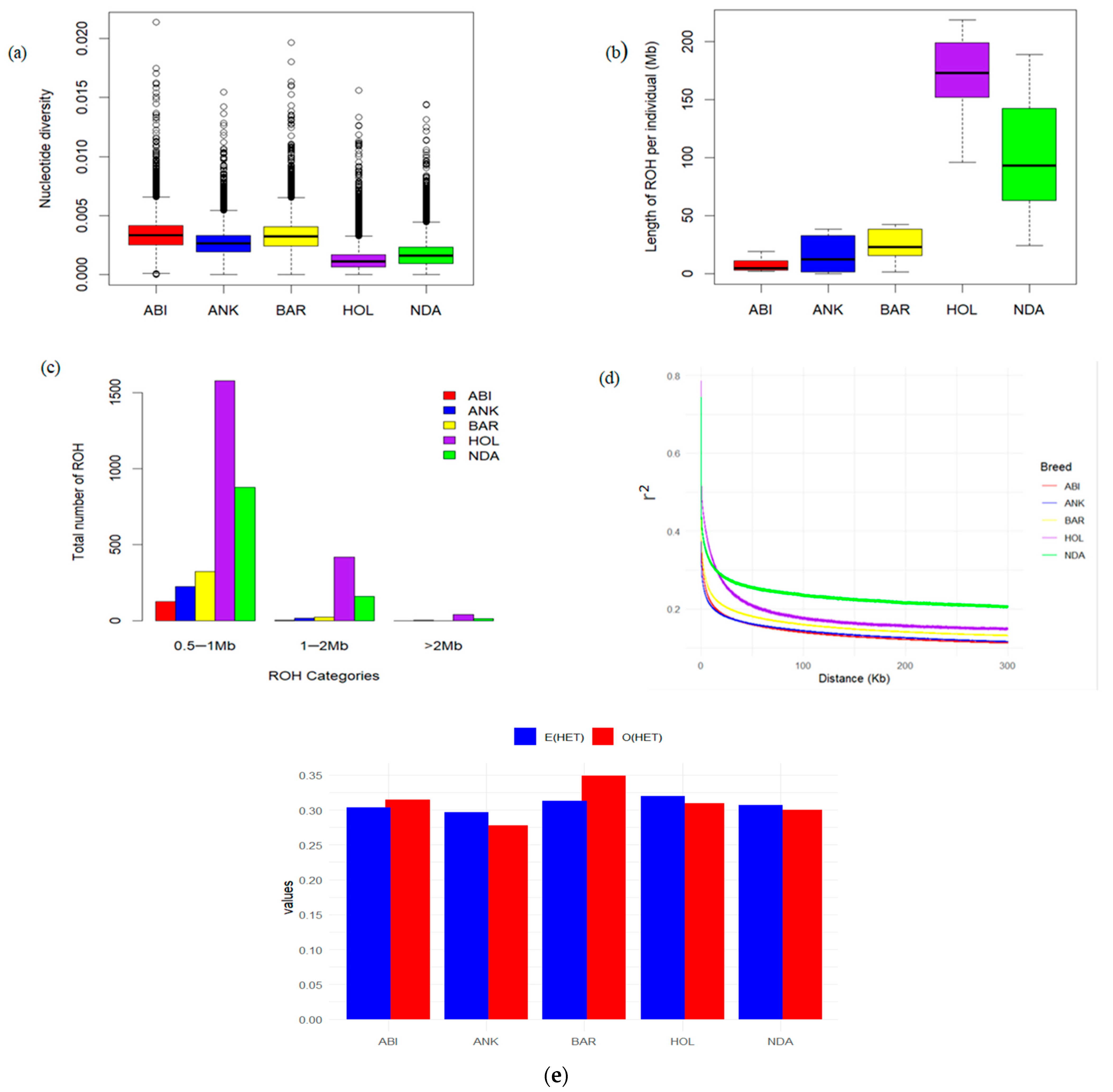
3.3. Patterns of Genomic Diversity
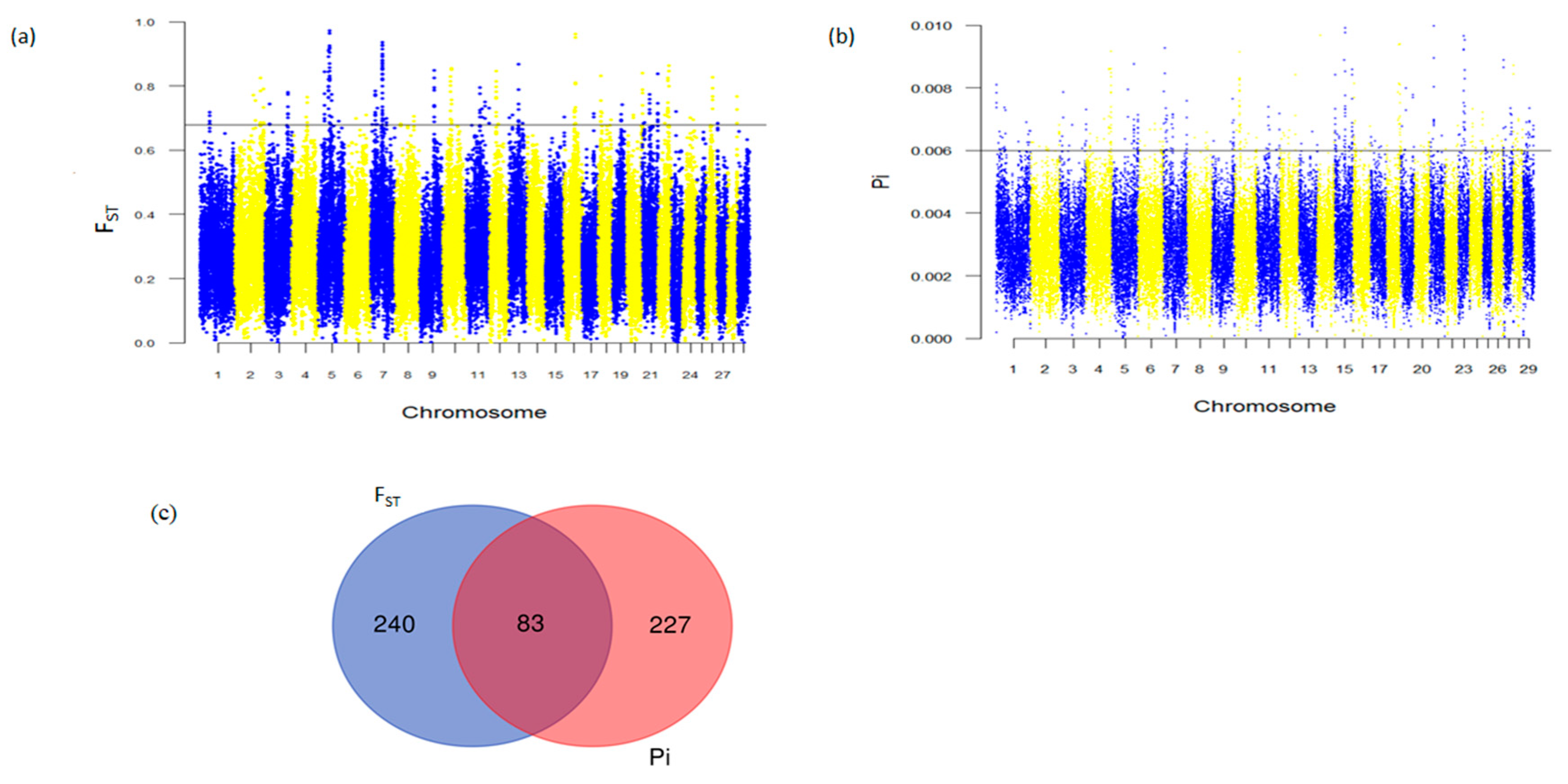
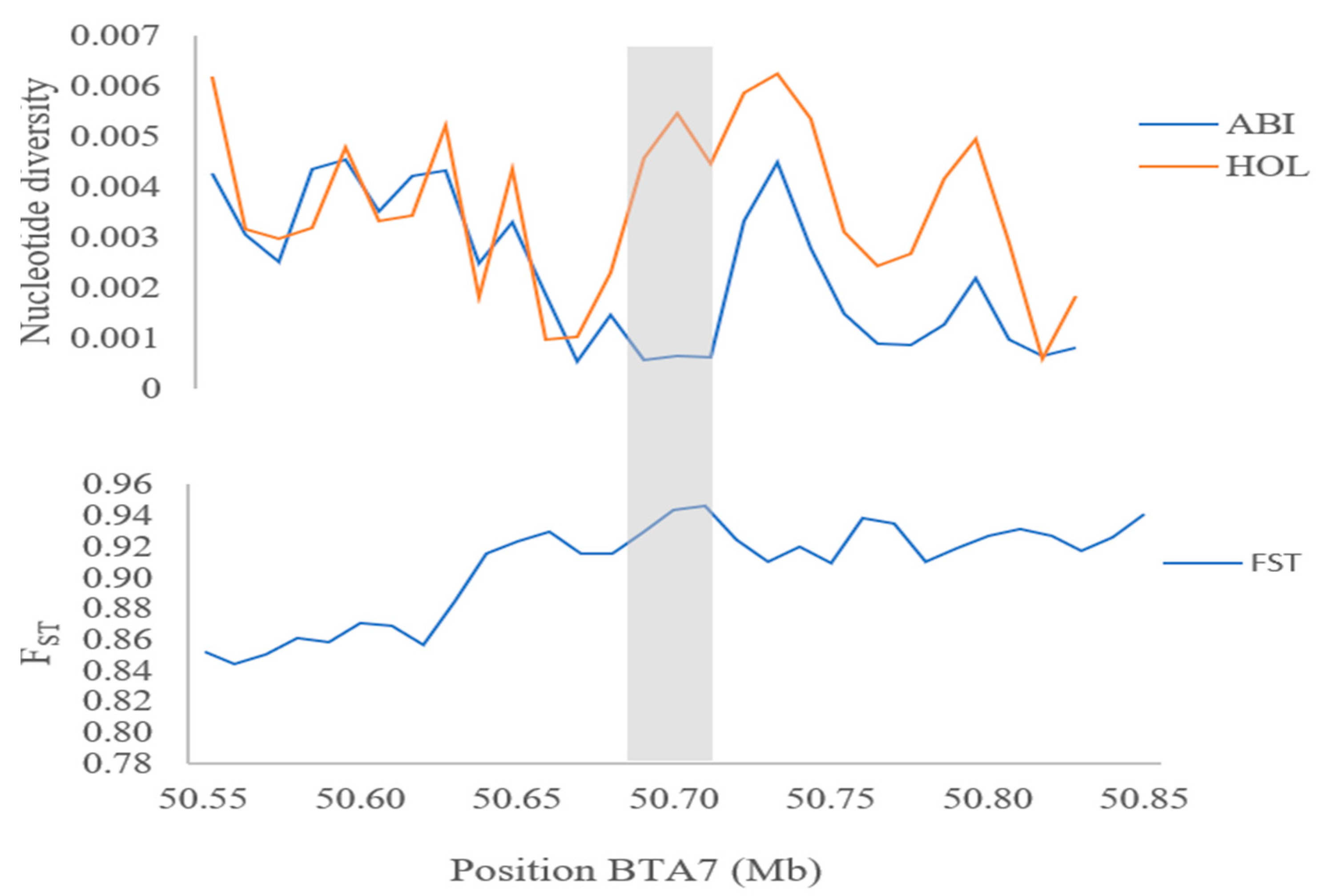
3.4. Genome-Wide Selective Sweeps
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schneider, H.K. The subsistence role of cattle among the Pakot and in East Africa. Am. Anthropol. 1957, 59, 278–300. [Google Scholar] [CrossRef]
- Di Lernia, S.; Tafuri, M.A.; Gallinaro, M.; Alhaique, F.; Balasse, M.; Cavorsi, L.; Fullagar, P.D.; Mercuri, A.M.; Monaco, A.; Perego, A.; et al. Inside the “African cattle complex”: Animal burials in the Holocene central Sahara. PLoS ONE 2013, 8, e56879. [Google Scholar] [CrossRef] [PubMed]
- Rege, J.E.O. The state of African cattle genetic resources I. Classification framework and identification of threatened and extinct breeds. Anim. Genet. Res. Inf. Bull. 1999, 25, 1–26. [Google Scholar] [CrossRef]
- Mwai, O.; Hanotte, O.; Kwon, Y.J.; Cho, S. African indigenous cattle: Unique genetic resources in a rapidly changing world. Asian-Australas. J. Anim. Sci. 2015, 28, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hanotte, O.; Mwai, O.A.; Dessie, T.; Bashir, S.; Diallo, B.; Agaba, M.; Kim, K.; Kwak, W.; Sung, S.; et al. The genome landscape of indigenous African cattle. Genome Biol. 2017, 18, 34. [Google Scholar] [CrossRef]
- Taye, M.; Lee, W.; Caetano-Anolles, K.; Dessie, T.; Hanotte, O.; Mwai, O.A.; Kemp, S.; Cho, S.; Oh, S.J.; Lee, H.K.; et al. Whole genome detection of signature of positive selection in African cattle reveals selection for thermotolerance. Anim. Sci. J. 2017, 88, 1889–1901. [Google Scholar] [CrossRef]
- Johnsson, M. Integrating selection mapping with genetic mapping and functional genomics. Front. Genet. 2018, 9, 603. [Google Scholar] [CrossRef]
- Tijjani, A.; Salim, B.; da Silva, M.V.; Eltahir, H.A.; Musa, T.H.; Marshall, K.; Hanotte, O.; Musa, H.H. Genomic signatures for drylands adaptation at gene-rich regions in African zebu cattle. Genomics 2022, 114, 110423. [Google Scholar] [CrossRef]
- Terefe, E.; Belay, G.; Tijjani, A.; Han, J.; Hanotte, O. Whole Genome Resequencing Reveals Genetic Diversity and Selection Signatures of Ethiopian Indigenous Cattle Adapted to Local Environments. Diversity 2023, 15, 540. [Google Scholar] [CrossRef]
- Kim, S.J.; Ka, S.; Ha, J.W.; Kim, J.; Yoo, D.; Kim, K.; Lee, H.K.; Lim, D.; Cho, S.; Hanotte, O.; et al. Cattle genome-wide analysis reveals genetic signatures in trypanotolerant N’Dama. BMC Genom. 2017, 18, 371. [Google Scholar] [CrossRef]
- Ayalew, W.; Wu, X.; Tarekegn, G.M.; Min, C.; Liang, C.; Tessema, T.S.; Ping, Y. Signatures of positive selection for local adaptation of African Native Cattle populations: A review. J. Integer. Agri. 2023, 22, 1967–1984. [Google Scholar] [CrossRef]
- Domestic Animal Diversity Information System (DADIS). Number of Breeds by Species and Country. Available online: http://dad.fao.org/ (accessed on 20 June 2021).
- Alberro, M.; Haile-Mariam, S. The indigenous cattle of Ethiopia Part I. FAO World Anim. Rev. 1982, 41, 2–10. [Google Scholar]
- Minuye, N.; Abebe, G.; Dessie, T. On-farm description and status of Nuer (Abigar) cattle breed in Gambella Regional State, Ethiopia. Int. J. Biodiv. Cons. 2018, 10, 292–302. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; DePristo, M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. Pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Res. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.; Xu, J.-Y.; He, W.-M.; Yang, T.L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Y.; Qi, X.; Cheng, H.; Chen, N.; Ahmed, Z.; Chen, Q.; Lei, C.; Yang, X. Genome-wide analysis emancipates genomic diversity and signature of selection in Altay white-headed cattle of Xinjiang, China. Front. Genet. 2023, 14, 1144249. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Ben-Jemaa, S.; Adam, G.; Boussaha, M.; Bardou, P.; Klopp, C.; Mandonnet, N.; Naves, M. Whole genome sequencing reveals signals of adaptive admixture in Creole cattle. Sci. Rep. 2023, 13, 12155. [Google Scholar] [CrossRef]
- Fujimoto, M.; Oshima, K.; Shinkawa, T.; Wang, B.B.; Inouye, S.; Hayashida, N.; Takii, R.; Nakai, A. Analysis of HSF4 binding regions reveals its necessity for gene regulation during development and heat shock response in mouse lenses. J. Biol. Chem. 2008, 31, 29961–29970. [Google Scholar] [CrossRef]
- Li, Y.; Zou, S.; Ding, H.; Hao, N.; Huang, Y.; Tang, J.; Cheng, J.; Feng, S.; Li, J.; Wang, X.; et al. Low expression of sirtuin 1 in the dairy cows with mild fatty liver alters hepatic lipid metabolism. Animals 2020, 10, 560. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and its roles in inflammation. Front. Immunol. 2022, 13, 831168. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, I.A.; Khatkar, M.S.; Thomson, P.C.; Raadsma, H.W. A meta-assembly of selection signatures in cattle. PLoS ONE 2016, 11, e0153013. [Google Scholar] [CrossRef] [PubMed]
- Onzima, R.B.; Upadhyay, M.R.; Doekes, H.P.; Brito, L.F.; Bosse, M.; Kanis, E.; Groenen, M.A.M.; Crooijmans, R. Genome-Wide Characterization of Selection Signatures and Runs of Homozygosity in Ugandan Goat Breeds. Front. Genet. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Bahbahani, H.; Tijjani, A.; Mukasa, C.; Wragg, D.; Almathen, F.; Nash, O.; Akpa, G.N.; Mbole-Kariuki, M.; Malla, S.; Woolhouse, M.; et al. Signatures of selection for environmental adaptation and Zebu × Taurine hybrid fitness in East African Shorthorn zebu. Front. Genet. 2017, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Nanaei, H.A.; Qanatqestani, M.D.; Esmailizadeh, A. Whole genome resequencing reveals selection signatures associated with milk production traits in African Kenana dairy zebu cattle. Genomics 2020, 112, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef]
- Fanta, M. Physiological adaptation of Holstein Frisian dairy cattle in Ethiopia: Review article. J. Biol. Agric. Health 2017, 7, 67–78. [Google Scholar]
- Leite, J.H.G.M.; Silva, R.G.; da Silva, W.S.T.; da Silva, W.E.; Paiva, R.D.M.; Sousa, J.E.R.; Façanha, D.A.E. Locally adapted Brazilian ewes with different coat colors maintain homeothermy during the year in an equatorial semiarid environment. Int. J. Biometeorol. 2018, 62, 1635–1644. [Google Scholar] [CrossRef]
- Finch, V.A.; Bennett, I.L.; Holmes, C.R. Coat colour in cattle: Effect on thermal balance, behaviour and growth, and relationship with coat type. J. Agri. Sci. 1984, 102, 141–147. [Google Scholar] [CrossRef]
- Gaughan, J.B.; Sejian, V.; Mader, T.L.; Dunshea, F.R. Adaptation strategies: Ruminants. Anim. Front. 2019, 9, 47–53. [Google Scholar] [CrossRef]
- Barsh, G.; Gunn, T.; He, L.; Schlossman, S.; Duke-Cohan, J. Biochemical and genetic studies of pigment-type switching. Pigment Cell Res. 2000, 13, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Saif, R.; Jagannathan, V.; Schmocker, C.; Zeindler, F.; Bangerter, E.; Herren, U.; Posantzis, D.; Bulut, Z.; Ammann, P.; et al. Selection signatures in goats reveal copy number variants underlying breed-defining coat color phenotypes. PLoS Genet. 2019, 15, e1008536. [Google Scholar] [CrossRef]
- Norris, B.J.; Whan, V.A. A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res. 2008, 18, 1282–1293. [Google Scholar] [CrossRef]
- Trigo, B.B.; Utsunomiya, A.T.; Fortunato, A.A.; Milanesi, M.; Torrecilha, R.B.; Lamb, H.; Nguyen, L.; Ross, E.M.; Hayes, B.; Padula, R.; et al. Variants at the ASIP locus contribute to coat color darkening in Nellore cattle. Genet. Selec. Evol. 2021, 53, 40. [Google Scholar] [CrossRef] [PubMed]
- Fontanesi, L.; Forestier, L.; Allain, D.; Scotti, E.; Beretti, F.; Deretz-Picoulet, S.; Pecchioli, E.; Vernesi, C.; Robinson, T.J.; Malaney, J.L.; et al. Characterization of the rabbit agouti signaling protein (ASIP) gene: Transcripts and phylogenetic analyses and identification of the causative mutation of the nonagouti black coat colour. Genomics 2010, 95, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Ivell, R.; Liu, X.; Janowski, D.; Anand-Ivell, R. Relaxin-family peptide receptors 1 and 2 are fully functional in the bovine. Front. Physiol. 2017, 8, 359. [Google Scholar] [CrossRef]
- Pan, Z.; Li, S.; Liu, Q.; Wang, Z.; Zhou, Z.; Di, R.; Miao, B.; Hu, W.; Wang, X.; Hu, X.; et al. Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. GigaScience 2018, 7, giy019. [Google Scholar] [CrossRef]
- Picard, K.; Thomas, D.W.; Festa-Bianchet, M.; Belleville, F.; Laneville, A. Differences in the thermal conductance of tropical and temperate bovid horns. Ecoscience 1999, 6, 148–158. [Google Scholar] [CrossRef]
- Langman, V.A.; Maloiy, G.M.O.; Schmidt-Nielsen, K.; Schroter, R.C. Nasal heat exchange in the giraffe and other large mammals. Respir. Physiol. 1979, 37, 325–333. [Google Scholar] [CrossRef]
- Fernandez-Guerrero, M.; Yakushiji-Kaminatsui, N.; Lopez-Delisle, L.; Zdral, S.; Darbellay, F.; Perez-Gomez, R.; Bolt, C.C.; Sanchez-Martin, M.A.; Duboule, D.; Ros, M.A. Mammalian-specific ectodermal enhancers control the expression of Hoxc genes in developing nails and hair follicles. Proc. Natl. Acad. Sci. USA 2020, 117, 30509–30519. [Google Scholar] [CrossRef]
- Alfonzo, E.P.M.; Barbosa Da Silva, M.V.G.; Dos Santos Daltro, D.; Stumpf, M.T.; Dalcin, V.C.; Kolling, G.; Fischer, V.; McManus, M. Relationship between physical attributes and heat stress in dairy cattle from different genetic groups. Int. J. Biometeorol. 2016, 60, 245–253. [Google Scholar] [CrossRef]
- Bahbahani, H.; Salim, B.; Almathen, F.; Al Enezi, F.; Mwacharo, J.M.; Hanotte, O. Signatures of positive selection in African Butana and Kenana dairy zebu cattle. PLoS ONE 2018, 13, e0190446. [Google Scholar] [CrossRef]
- Paguem, A.; Abanda, B.; Achukwi, M.D.; Baskaran, P.; Czemmel, S.; Renz, A.; Eisenbarth, A. Whole genome characterization of autochthonous Bos taurus brachyceros and introduced Bos indicus indicus cattle breeds in Cameroon regarding their adaptive phenotypic traits and pathogen resistance. BMC Genet. 2020, 21, 64. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Andreani, V.; Kapoor, T.; Herp, S.; Flach, H.; Duchniewicz, M.; and Grosschedl, R. MZB1 is a GRP94 cochaperone that enables proper immunoglobulin heavy chain biosynthesis upon ER stress. Genes Dev. 2014, 28, 1165–1178. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.N.; Ju, J.M.; Shabanova, A.; Li, Y.; Fang, R.N.; Yang, B.F. Mzb1 protects against myocardial infarction injury in mice via modulating mitochondrial function and alleviating inflammation. Acta Pharmacol. Sin. 2021, 42, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, H.C.; Engel, S.A.M.; Eck, P.K.; Welch, R.; Yeager, M.; Levine, M.; Siega-Riz, A.M.; Olshan, A.F.; Chanock, S.J. Genetic variation in the sodium-dependent vitamin C transporters, SLC23A1, and SLC23A2 and risk for preterm delivery. Am. J. Epidemiol. 2006, 163, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Maggini, S. Vitamin C and immune function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef]
- Ishikawa, H.; and Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Chinenov, Y.; Gupte, R.; Dobrovolna, J.; Flammer, J.R.; Liu, B.; Michelassi, F.E.; Rogatsky, I. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc. Natl. Acad. Sci. USA 2012, 109, 11776–11781. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Turek, I.; Meehan-Andrews, T.; Zacharias, A.; Irving, H. Analysis of interleukin-1 receptor associated kinase-3 (IRAK3) function in modulating expression of inflammatory markers in cell culture models: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0244570. [Google Scholar] [CrossRef]
- Tunalı, G.; Bedós, M.R.; Nagarajan, D.; Fridh, P.; Papakyriacou, I.; Mao, Y. IL-1 receptor–associated kinase-3 acts as an immune checkpoint in myeloid cells to limit cancer immunotherapy. J. Clin. Investig. 2023, 133, e161084. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abigar | Ankole | Barca | Holstein | NDama | |
|---|---|---|---|---|---|
| Abigar | |||||
| Ankole | 0.055 | ||||
| Barca | 0.062 | 0.122 | |||
| Holstein | 0.275 | 0.250 | 0.349 | ||
| NDama | 0.189 | 0.173 | 0.267 | 0.265 |
| Methods | BTA | Gene | Ensemble ID | Summary of Gene Function | References |
|---|---|---|---|---|---|
| Pi | 13 | ASIP | ENSBTAG00000034077 | Heat tolerance/coat color | [29] |
| 18 | HSF4 | ENSBTAG0000000354 | Heat tolerance | [30] | |
| 28 | SIRT1 | ENSBTAG00000014023 | Oxidative stress response | [31,32] | |
| FST | 5 | GRIP1 | ENSBTAG00000033726 | Immune response | [31] |
| 7 | HSPA9 | ENSBTAG00000011419 | Immune response | [9,33] | |
| Both (Pi and FST) | 5 | IRAK3 | ENSBTAG00000007636 | Immune response | [8] |
| 5 | HOXC13 | ENSBTAG00000000923 | Heat tolerance | [6,32,34] | |
| 5 | WIF1 | ENSBTAG00000014758 | Immune response | [9,33] | |
| 7 | MZB1 | ENSBTAG00000038337 | Immune response | [9] | |
| 7 | STING1 | ENSBTAG00000002296 | Immune response | [9] | |
| 7 | DNAJC18 | ENSBTAG00000002286 | Heat tolerance | [9,35] | |
| 7 | SLC23A1 | ENSBTAG00000010798 | Oxidative stress response | [6] | |
| 12 | RXFP2 | ENSBTAG00000015132 | Heat tolerance, horn development | [31] | |
| 16 | FAAP20 | ENSBTAG00000014136 | Dryland adaptation (scarce feed and water supply) | [8] | |
| 16 | SKI | ENSBTAG00000038716 | Dryland adaptation (scarce feed and water supply) | [8] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayalew, W.; Wu, X.; Tarekegn, G.M.; Sisay Tessema, T.; Naboulsi, R.; Van Damme, R.; Bongcam-Rudloff, E.; Edea, Z.; Enquahone, S.; Yan, P. Whole-Genome Resequencing Reveals Selection Signatures of Abigar Cattle for Local Adaptation. Animals 2023, 13, 3269. https://doi.org/10.3390/ani13203269
Ayalew W, Wu X, Tarekegn GM, Sisay Tessema T, Naboulsi R, Van Damme R, Bongcam-Rudloff E, Edea Z, Enquahone S, Yan P. Whole-Genome Resequencing Reveals Selection Signatures of Abigar Cattle for Local Adaptation. Animals. 2023; 13(20):3269. https://doi.org/10.3390/ani13203269
Chicago/Turabian StyleAyalew, Wondossen, Xiaoyun Wu, Getinet Mekuriaw Tarekegn, Tesfaye Sisay Tessema, Rakan Naboulsi, Renaud Van Damme, Erik Bongcam-Rudloff, Zewdu Edea, Solomon Enquahone, and Ping Yan. 2023. "Whole-Genome Resequencing Reveals Selection Signatures of Abigar Cattle for Local Adaptation" Animals 13, no. 20: 3269. https://doi.org/10.3390/ani13203269
APA StyleAyalew, W., Wu, X., Tarekegn, G. M., Sisay Tessema, T., Naboulsi, R., Van Damme, R., Bongcam-Rudloff, E., Edea, Z., Enquahone, S., & Yan, P. (2023). Whole-Genome Resequencing Reveals Selection Signatures of Abigar Cattle for Local Adaptation. Animals, 13(20), 3269. https://doi.org/10.3390/ani13203269