
Characterization of Lung Microbiomes in Pneumonic Hu Sheep Using Culture Technique and 16S rRNA Gene Sequencing

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Sample Collection
2.3. Microbial Isolation and Identification
2.4. DNA Extraction, PCR and Sequencing
2.5. Sequence Analysis
2.6. Statistical Analysis
3. Results
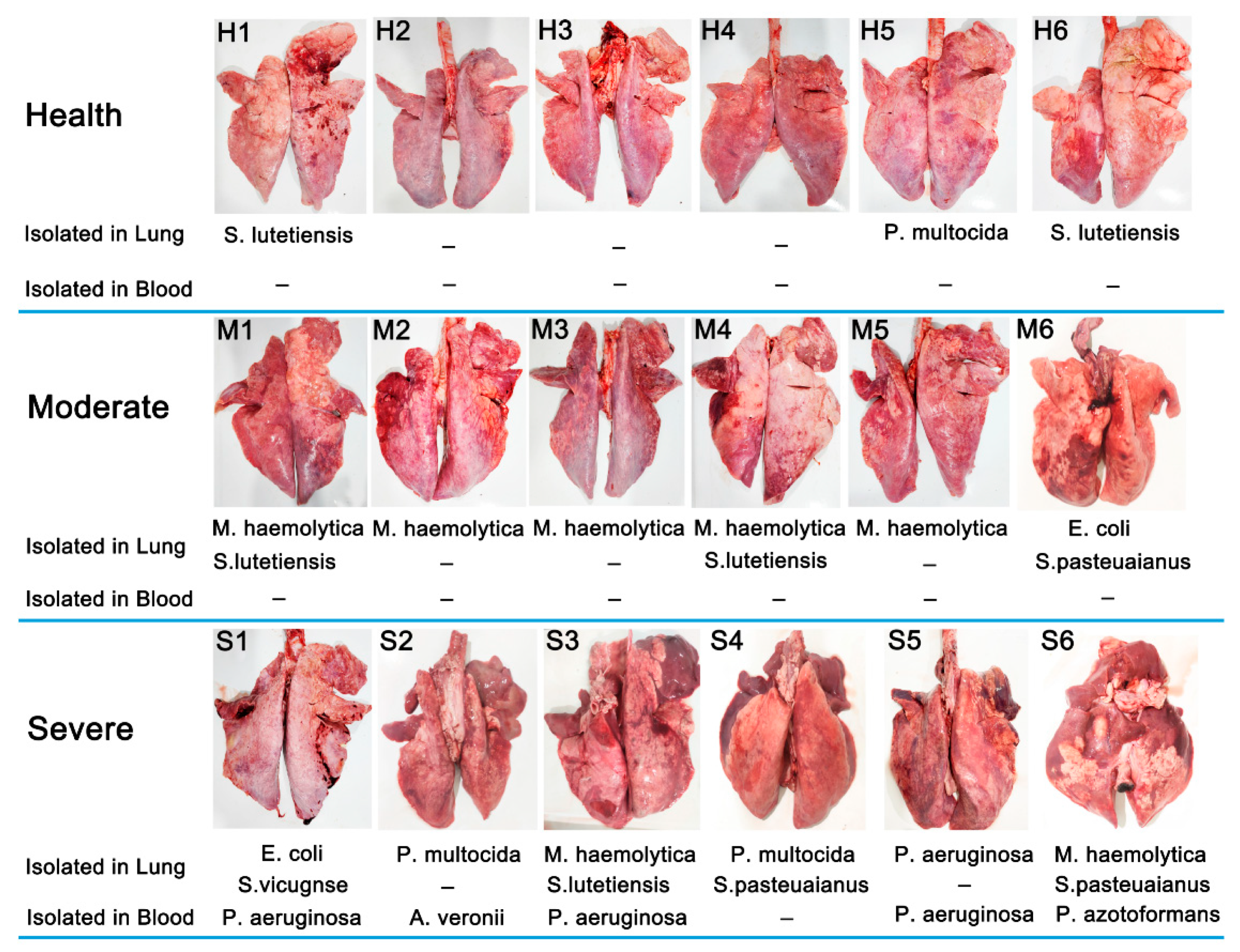
3.1. Outcome of Bacterial Isolation
3.2. 16S rRNA Sequencing Analysis
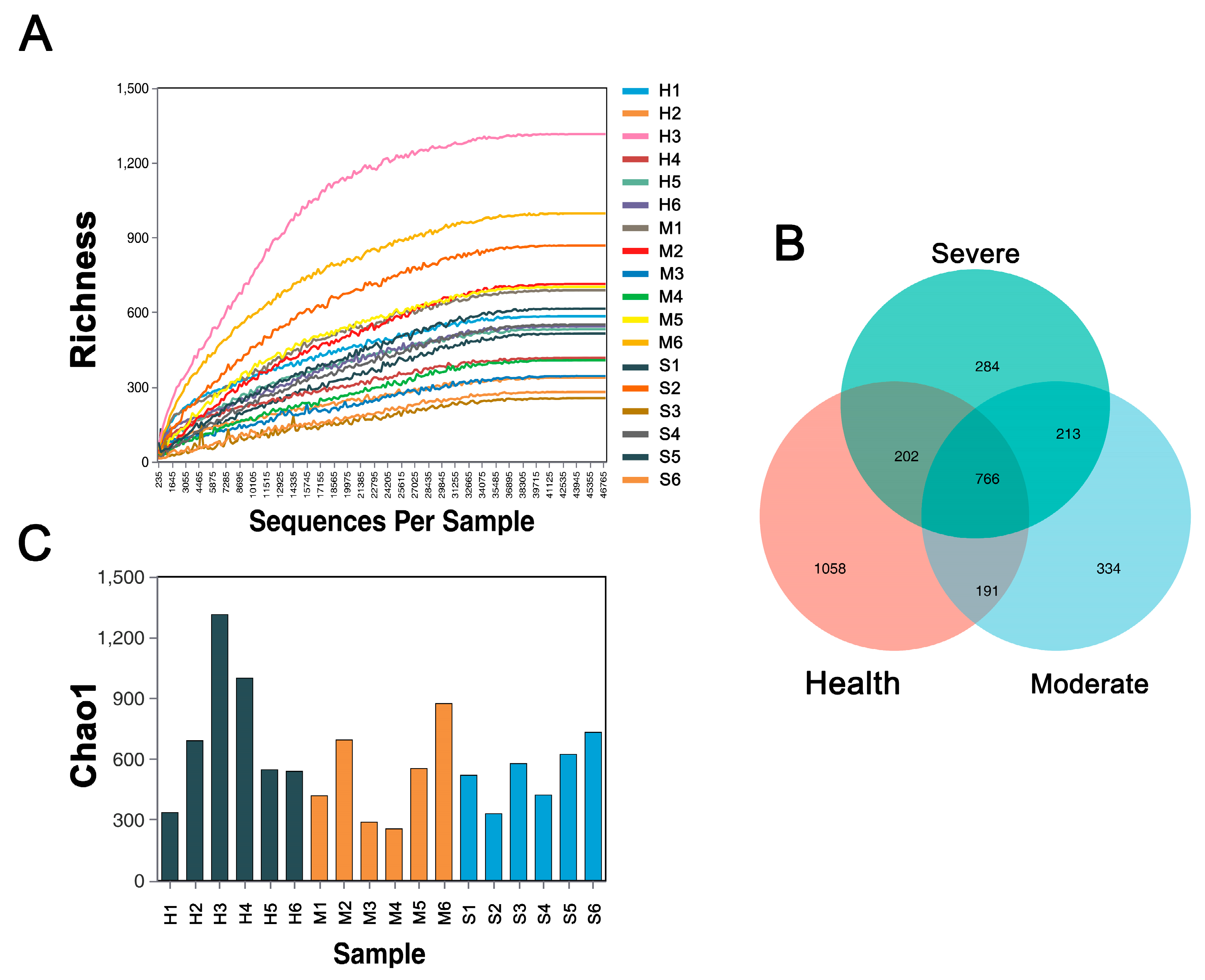
3.3. Analysis of Microbial Diversity in the Lungs
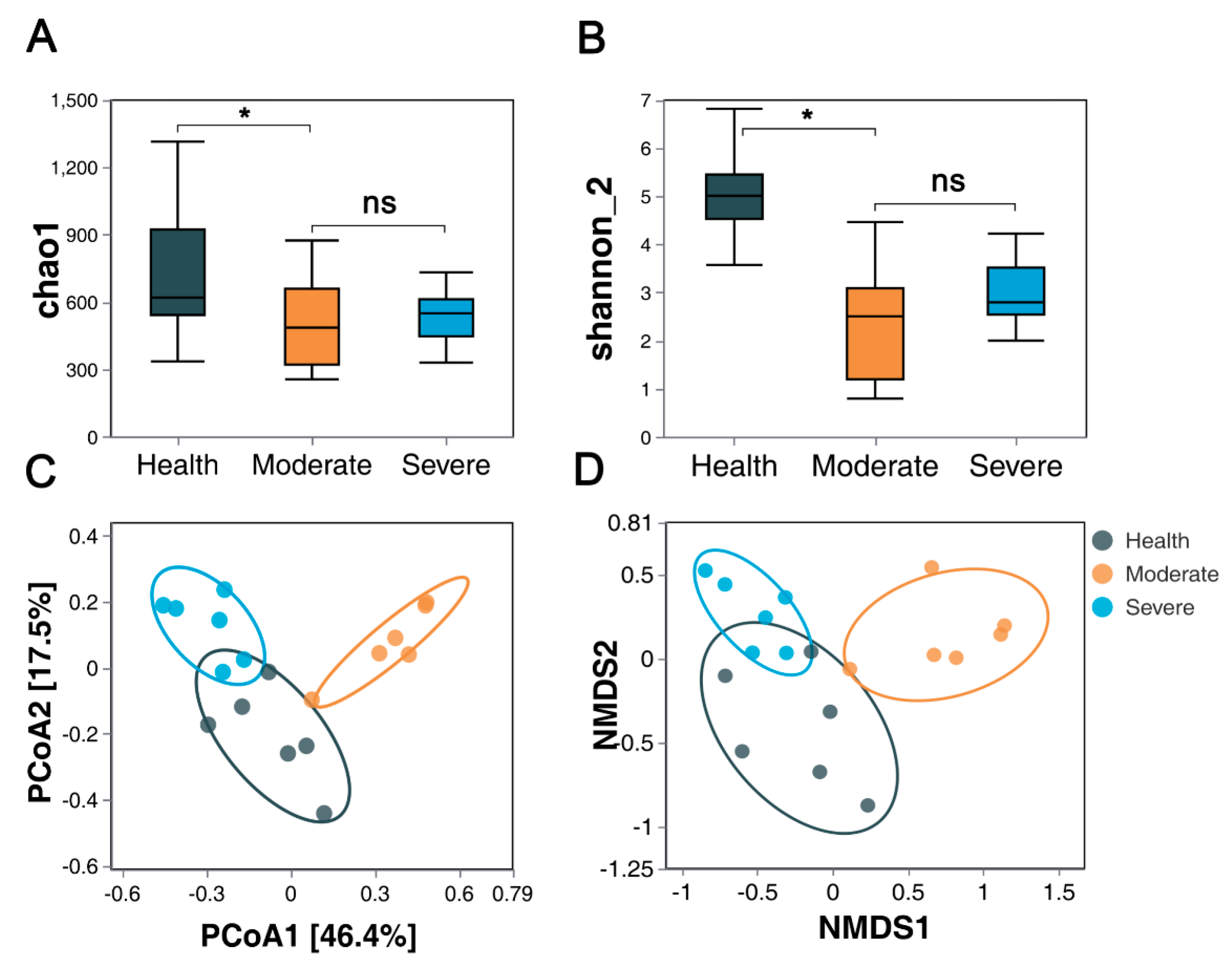
3.3.1. Bacterial Community α-Diversity
3.3.2. Bacterial Community β-Diversity
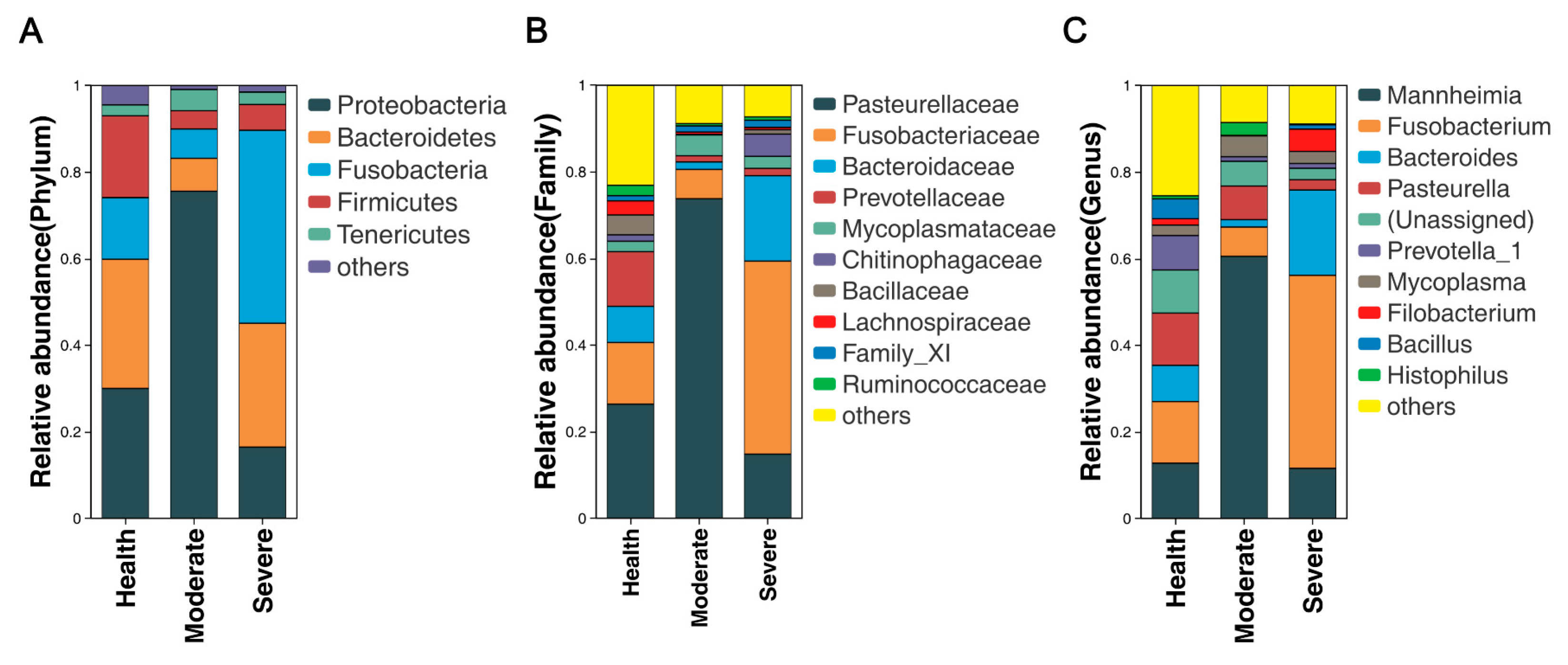
3.4. Bacterial Community Composition at Various Taxonomic Levels in the Lungs
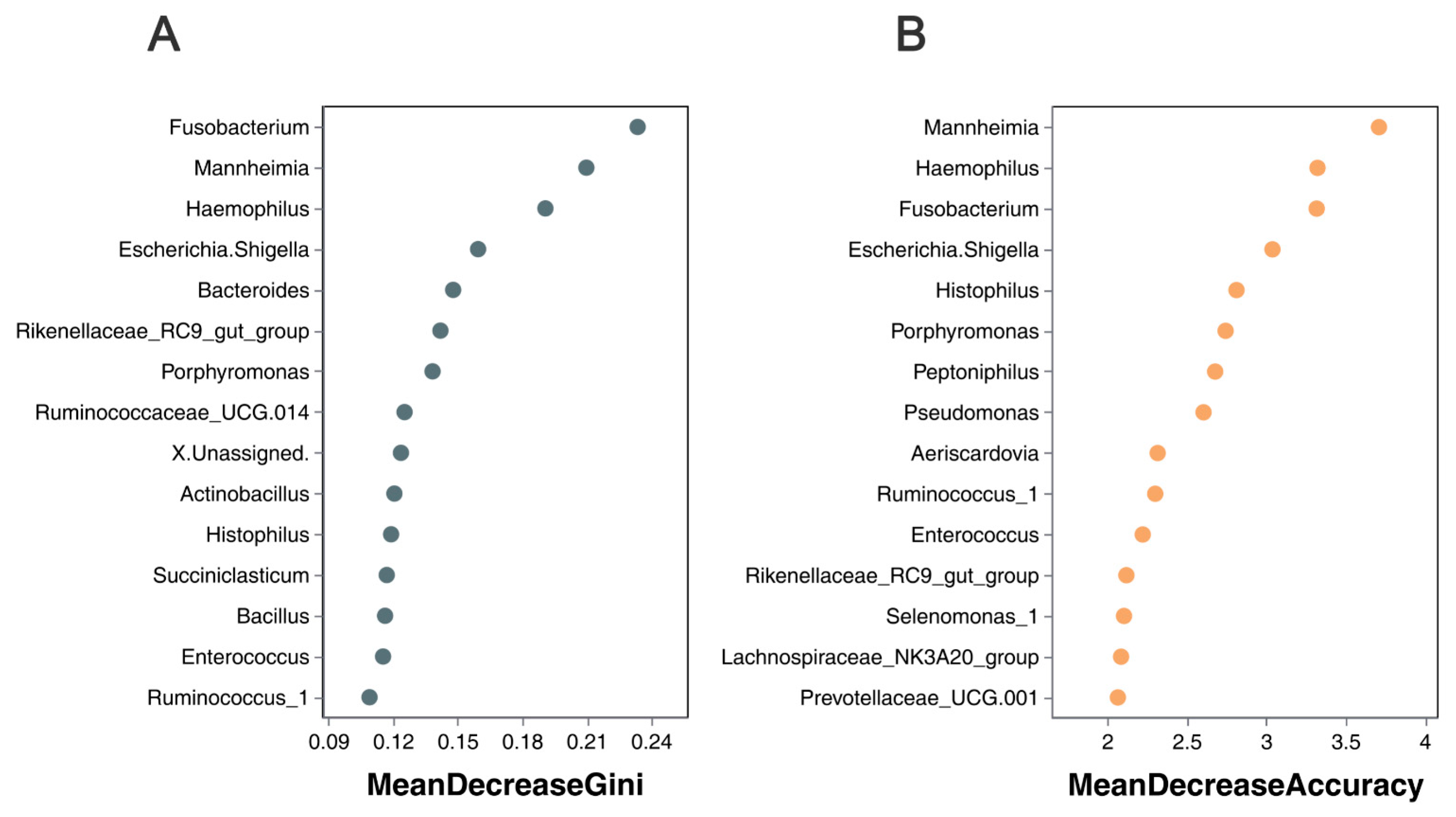
3.5. Overview and Comparison of Lung Microbiota at the Genus Level
3.6. Comparison of Microbiotas in Lungs
3.7. Correlation of Lung Microbiota in Sheep with Severity of Pneumonia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tabatabaei, M.; Abdollahi, A.F. Isolation and identification of M. haemolytica by culture and polymerase chain reaction from sheep’s pulmonary samples in Shiraz, Iran. Vet. World 2018, 11, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Mekibib, B.; Mikir, T.; Fekadu, A.; Abebe, R. Prevalence of Pneumonia in Sheep and Goats Slaughtered at Elfora Bishoftu Export Abattoir, Ethiopia: A Pathological Investigation. J. Vet. Med. 2019, 5, 5169040. [Google Scholar] [CrossRef] [PubMed]
- Grossman, P.C.; Schneider, D.A.; Herndon, D.R.; Knowles, D.P.; Highland, M.A. Differential pulmonary immunopathology of domestic sheep (Ovis aries) and bighorn sheep (Ovis canadensis) with Mycoplasma ovipneumoniae infection: A retrospective study. Comp. Immunol. Microbiol. Infect. Dis. 2021, 76, 101641. [Google Scholar] [CrossRef] [PubMed]
- Getnet, K.; Abera, B.; Getie, H.; Molla, W. Serotyping and Seroprevalence of M. haemolytica, Pasteurella multocida, and Bibersteinia trehalosi and Assessment of Determinants of Ovine Pasteurellosis in West Amhara Sub-region, Ethiopia. Front. Vet. Sci. 2022, 9, 866206. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.; Calderón Bernal, J.M.; Torre-Fuentes, L.; Hernández, M.; Jimenez, C.E.P.; Domínguez, L.; Fernández-Garayzábal, J.F.; Vela, A.I.; Cid, D. Antimicrobial Susceptibility and Resistance Mechanisms in Mannheimia haemolytica Isolates from Sheep at Slaughter. Animals 2023, 13, 1991. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, L.E.; St. George, T.D.; Sullivan, N.D.; Horsfall, N. Isolation, propagation, and characterization studies of an ovine Mycoplasma responsible for proliferative interstitial pneumonia. Cornell Vet. 1972, 62, 654–679. [Google Scholar] [PubMed]
- Manlove, K.; Branan, M.; Baker, K.; Bradway, D.; Cassirer, E.F.; Marshall, K.L.; Miller, R.S.; Sweeney, S.; Cross, P.C.; Besser, T.E. Risk factors and productivity losses associated with Mycoplasma ovipneumoniae infection in United States domestic sheep operations. Prev. Vet. Med. 2019, 168, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, N.C.; Sá Guimarães, A.M.; Timenetsky, J.; Clouser, S.; Gregory, L.; Ganda, E. The first Mycoplasma ovipneumoniae recovered from a sheep with respiratory disease in Brazil—Draft genome and genomic analysis. Vet. Res. Commun. 2022, 46, 1311–1318. [Google Scholar] [CrossRef]
- Windsor, P.A. Anaemia in Lambs Caused by Mycoplasma ovis: Global and Australian Perspectives. Animals 2022, 12, 1372. [Google Scholar] [CrossRef]
- Barbour, E.K.; Nabbut, N.H.; Hamadeh, S.K.; Al-Nakhli, H.M. Bacterial identity and characteristics in healthy and unhealthy respiratory tracts of sheep and calves. Vet. Res. Commun. 1997, 21, 421–430. [Google Scholar] [CrossRef]
- Dassanayake, R.P.; Shanthalingam, S.; Herndon, C.N.; Subramaniam, R.; Lawrence, P.K.; Bavananthasivam, J.; Cassirer, E.F.; Haldorson, G.J.; Foreyt, W.J.; Rurangirwa, F.R.; et al. Mycoplasma ovipneumoniae can predispose bighorn sheep to fatal M. haemolytica pneumonia. Vet. Microbiol. 2010, 145, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bernasconi, E.; Koutsokera, A.; Wurlod, D.; Tripathi, V.; Bonilla-Rosso, G. A prevalent and culturable microbiota links ecological balance to clinical stability of the human lung after transplantation. Nat. Commun. 2021, 12, 2126. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, L.W.; Wang, Z.; Ma, T.W.; Deng, M.T.; Wang, F.; Zhang, Y.L. Energy and protein requirements for maintenance of Hu sheep during pregnancy. J. Int. Agric. 2018, 17, 173–183. [Google Scholar] [CrossRef]
- Miao, Y.Q.; Zhao, X.L.; Adam, F.E.A.; Xie, Q.F.; Feng, H.; Ding, J.R.; Bai, X.; Wang, J.; Yang, Z.Q. Isolation and Identification of Aeromonas veronii in Sheep with Fatal Infection in China: A Case Report. Microorganisms 2023, 11, 333. [Google Scholar] [CrossRef]
- Glendinning, L.; Collie, D.; Wright, S.; Rutherford, K.M.D.; McLachlan, G. Comparing microbiotas in the upper aerodigestive and lower respiratory tracts of lambs. Microbiome 2017, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Glendinning, L.; Wright, S.; Pollock, J.; Tennant, P.; Collie, D.; McLachlan, G. Variability of the Sheep Lung Microbiota. Appl. Environ. Microbiol. 2016, 82, 3225–3238. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Sung, J.Y.; Yong, D.; Chun, J.; Kim, S.Y.; Song, J.H.; Chung, K.S.; Kim, E.Y. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 2016, 102, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; O’Brien, J.L.; Ajami, N.J.; Scheurer, M.E.; Amirian, E.S.; Armstrong, G.; Tsavachidis, S.; Thrift, A.P.; Jiao, L.; Wong, M.C.; et al. Lung tissue microbial profile in lung cancer is distinct from emphysema. Am. J. Cancer Res. 2018, 8, 1775–1787. [Google Scholar]
- Scheerlinck, J.P.; Snibson, K.J.; Bowles, V.M.; Sutton, P. Biomedical applications of sheep models: From asthma to vaccines. Trends Biotechnol. 2008, 26, 259–266. [Google Scholar] [CrossRef]
- McCarron, A.; Parsons, D.; Donnelley, M. Animal and Cell Culture Models for Cystic Fibrosis: Which Model Is Right for Your Application? Am. J. Pathol. 2021, 191, 228–242. [Google Scholar] [CrossRef]
- Xu, L.Q.; Yang, J.; Liang, W.; Chen, J.; Sun, Z.; Zhang, Q.; Liu, X.; Qiao, F.; Li, J. LDMD: A database of microbes in human lung disease. Front. Microbiol. 2023, 13, 1085079. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Deng, X.; Yang, X.; Wang, J.; Li, T.; Hua, G.; Han, D.; Da, L.; Li, R.; Rong, W.; et al. Characteristics of Bacterial Microbiota in Different Intestinal Segments of Aohan Fine-Wool Sheep. Front. Microbiol. 2022, 13, 874536. [Google Scholar] [CrossRef] [PubMed]
- Segal, L.N.; Clemente, J.C.; Tsay, J.; Koralov, S.; Keller, B.; Wu, B.; Li, Y.; Shen, N.; Ghedin, E.; Morris, A.; et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol. 2016, 1, 16031. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Villot, C.; Renaud, D.; Skidmore, A.; Chevaux, E.; Steele, M.; Guan, L.L. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: Prediction of diarrhea. ISME J. 2020, 14, 2223–2235. [Google Scholar] [CrossRef] [PubMed]
- Carney, S.M.; Clemente, J.C.; Cox, M.J.; Dickson, R.P.; Huang, Y.J.; Kitsios, G.D.; Kloepfer, K.M.; Leung, J.M.; LeVan, T.D.; Molyneaux, P.L.; et al. Methods in Lung Microbiome Research. Am. J. Respir. Cell Mol. Biol. 2020, 62, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.M.; Hinkle, K.J.; McDonald, R.A.; Brown, C.A.; Falkowski, N.R.; Huffnagle, G.B.; Dickson, R.P. Whole lung tissue is the preferred sampling method for amplicon-based characterization of murine lung microbiota. Microbiome 2021, 9, 99. [Google Scholar] [CrossRef]
- Kernaghan, S.; Bujold, A.R.; MacInnes, J.I. The microbiome of the soft palate of swine. Anim. Health Res. Rev. 2012, 13, 110–120. [Google Scholar] [CrossRef]
- Kiley, J.P. Advancing respiratory research. Chest 2011, 140, 497–501. [Google Scholar] [CrossRef][Green Version]
- Zimmermann, A.; Knecht, H.; Häsler, R.; Zissel, G.; Gaede, K.I.; Hofmann, S.; Nebel, A.; Müller-Quernheim, J.; Schreiber, S.; Fischer, A. Atopobium and Fusobacterium as novel candidates for sarcoidosis-associated microbiota. Eur. Respir. J. 2017, 50, 1600746. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Shariff, M.; Chaturvedi, G.; Sharma, A.; Goel, N.; Yadav, M.; Mortensen, M.S.; Sørensen, S.J.; Mukerji, M.; Chauhan, N.S. Comparative analysis of the alveolar microbiome in COPD, ECOPD, Sarcoidosis, and ILD patients to identify respiratory illnesses specific microbial signatures. Sci. Rep. 2021, 11, 3963. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, Y.; Li, J.; Tan, Y.; An, T.; Zhuo, M.; Pan, Z.; Ma, M.; Jia, B.; Zhang, H.; et al. Characterization of Lung and Oral Microbiomes in Lung Cancer Patients Using Culturomics and 16S rRNA Gene Sequencing. Microbiol. Spectr. 2023, 11, e0031423. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhang, M.; Tong, X.; Chen, J.; Yan, G.; Fang, S.; Guo, Y.; Yang, B.; Xiao, S.; Chen, C.; et al. Microbial communities in swine lungs and their association with lung lesions. Microb. Biotechnol. 2019, 12, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Wang, Y.; Li, Z.; Qamar, H.; Mehmood, K.; Zhang, L.; Liu, J.; Zhang, H.; Li, J. Probiotics isolated from yaks improves the growth performance, antioxidant activity, and cytokines related to immunity and inflammation in mice. Microb. Cell Fact. 2019, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.; Earley, B.; Cormican, P.; Murray, G.; Kenny, D.A.; Waters, S.M.; McGee, M.; Kelly, A.K.; McCabe, M.S. Illumina MiSeq 16S amplicon sequence analysis of bovine respiratory disease associated bacteria in lung and mediastinal lymph node tissue. BMC Vet. Res. 2017, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Shanthalingam, S.; Narayanan, S.; Batra, S.A.; Jegarubee, B.; Srikumaran, S. Fusobacterium necrophorum in North American Bighorn Sheep (Ovis canadensis) Pneumonia. J. Wildl. Dis. 2016, 52, 616–620. [Google Scholar] [CrossRef]
- Brogden, K.A.; Lehmkuhl, H.D.; Cutlip, R.C. Pasteurella haemolytica complicated respiratory infections in sheep and goats. Vet. Res. 1998, 29, 233–254. [Google Scholar]
- Dickson, R.P.; Singer, B.H.; Newstead, M.W.; Falkowski, N.R.; Erb-Downward, J.R.; Standiford, T.J.; Huffnagle, G.B. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 2016, 1, 16113. [Google Scholar] [CrossRef]
- Venkataraman, A.; Bassis, C.; Beck, J.M.; Young, V.B.; Curtis, J.L.; Huffnagle, G.; Schmidt, T.M. Application of a neutral community model to assess structuring of the human lung microbiome. MBio 2015, 6, e02284-14. [Google Scholar] [CrossRef]
- Gylswyk, N.O. Succiniclasticum ruminis gen. nov., sp. nov., a ruminal bacterium converting succinate to propionate as the sole energy-yielding mechanism. Int. J. Syst. Bacteriol. 1995, 45, 297–300. [Google Scholar] [CrossRef]
- Timsit, E.; McMullen, C.; Amat, S.; Alexander, T.W. Respiratory Bacterial Microbiota in Cattle: From Development to Modulation to Enhance Respiratory Health. The Veterinary clinics of North America. Vet. Clin. N. Am. Food Anim. Pract. 2020, 36, 297–320. [Google Scholar] [CrossRef]
- Amat, S.; Alexander, T.W.; Holman, D.B.; Schwinghamer, T.; Timsit, E. Intranasal Bacterial Therapeutics Reduce Colonization by the Respiratory Pathogen M. haemolytica in Dairy Calves. mSystems 2020, 5, e00629-19. [Google Scholar] [CrossRef]
- Cid, D.; Pinto, C.; Domínguez, L.; Vela, A.I.; Fernández-Garayzábal, J.F. Strength of association between isolation of Pasteurella multocida and consolidation lesions in ovine pneumonic pasteurellosis. Vet. Microbiol. 2020, 248, 108823. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, Y.; Zhao, X.; Lei, J.; Ding, J.; Feng, H.; Wu, K.; Liu, J.; Wang, C.; Ye, D.; Wang, X.; et al. Characterization of Lung Microbiomes in Pneumonic Hu Sheep Using Culture Technique and 16S rRNA Gene Sequencing. Animals 2023, 13, 2763. https://doi.org/10.3390/ani13172763
Miao Y, Zhao X, Lei J, Ding J, Feng H, Wu K, Liu J, Wang C, Ye D, Wang X, et al. Characterization of Lung Microbiomes in Pneumonic Hu Sheep Using Culture Technique and 16S rRNA Gene Sequencing. Animals. 2023; 13(17):2763. https://doi.org/10.3390/ani13172763
Chicago/Turabian StyleMiao, Yongqiang, Xueliang Zhao, Jianlin Lei, Jingru Ding, Hang Feng, Ke Wu, Jiaohu Liu, Chunyang Wang, Dongyang Ye, Xinglong Wang, and et al. 2023. "Characterization of Lung Microbiomes in Pneumonic Hu Sheep Using Culture Technique and 16S rRNA Gene Sequencing" Animals 13, no. 17: 2763. https://doi.org/10.3390/ani13172763
APA StyleMiao, Y., Zhao, X., Lei, J., Ding, J., Feng, H., Wu, K., Liu, J., Wang, C., Ye, D., Wang, X., Wang, J., & Yang, Z. (2023). Characterization of Lung Microbiomes in Pneumonic Hu Sheep Using Culture Technique and 16S rRNA Gene Sequencing. Animals, 13(17), 2763. https://doi.org/10.3390/ani13172763