
Comparative Transcriptomics Identify Key Pituitary Circular RNAs That Participate in Sheep (Ovis aries) Reproduction
 , ,
, ,  and
and 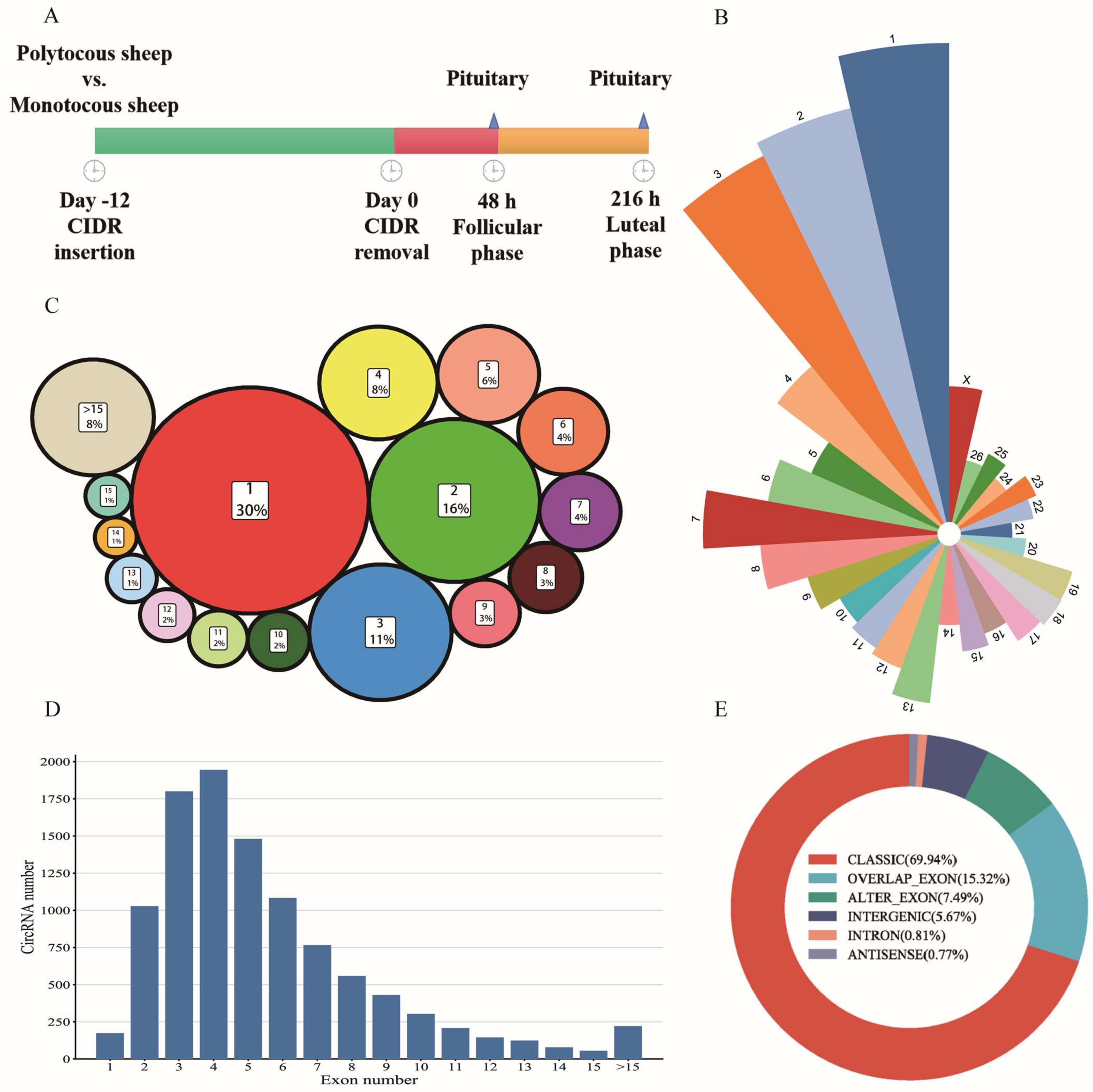{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics
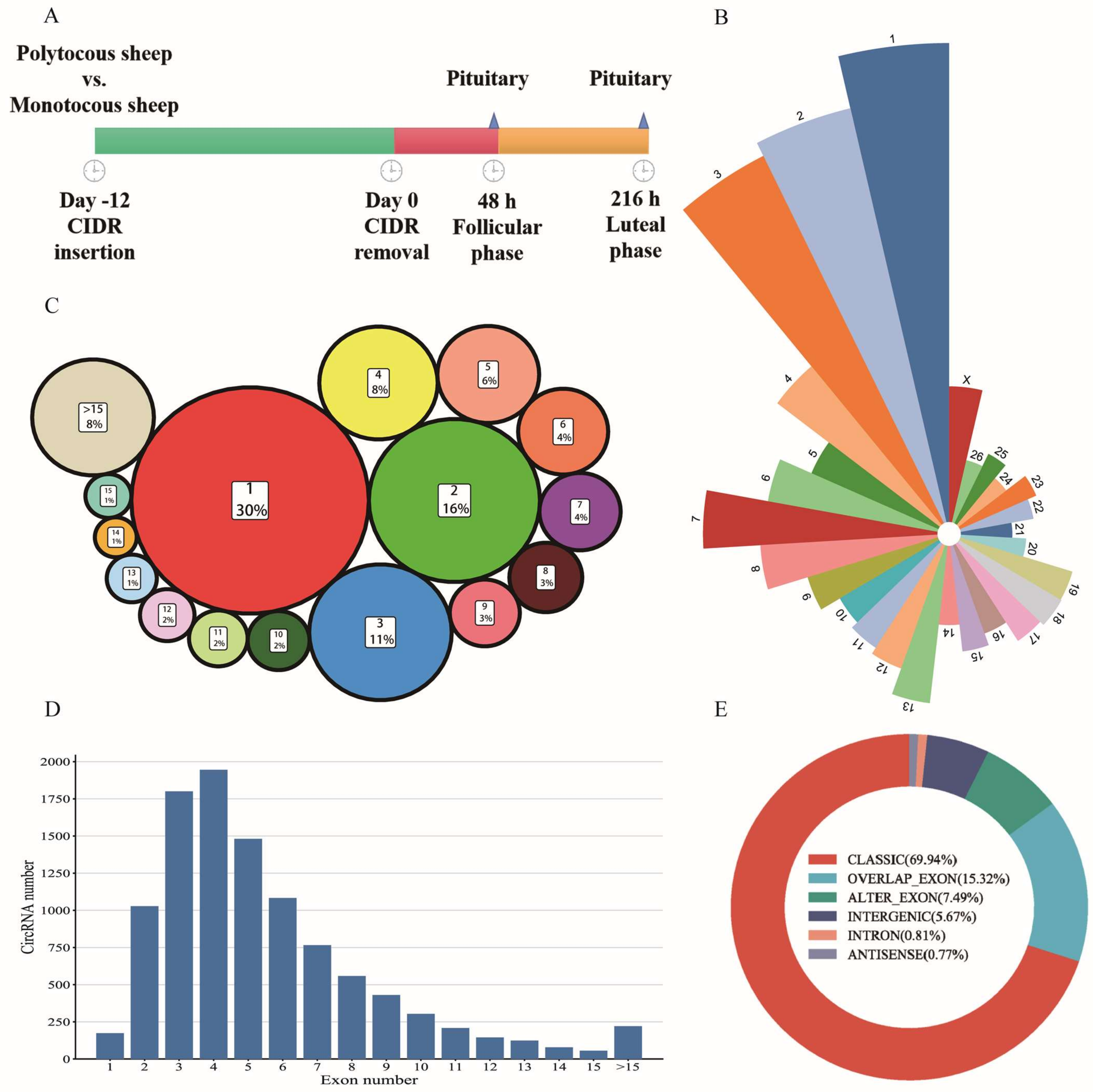
2.2. Animal Processing and Pituitary Collection
2.3. RNA Isolation, Library Construction, and Sequencing
2.4. Data Quality Control, Alignment, and CircRNA Identification
2.5. Differential Expression Analysis
2.6. Coding Potential Prediction of DE circRNAs
2.7. Bioinformatics Analysis of Source Genes of DE circRNAs
2.8. Prediction of the Targeting miRNA of DE circRNAs
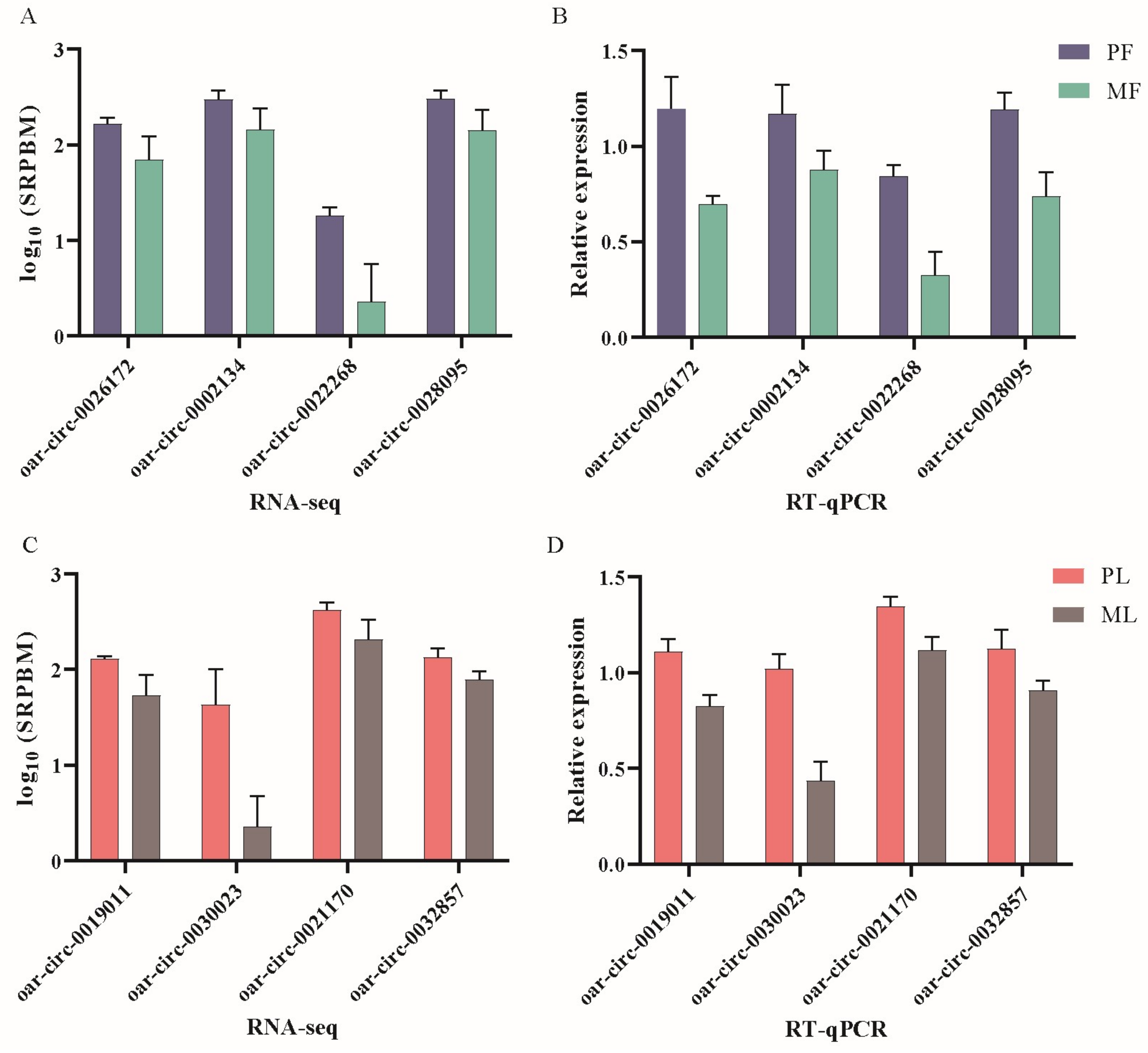
2.9. Validation of DE circRNAs
3. Results
3.1. Summary Statistics of RNA-seq Data and Characteristic of circRNAs in Pituitary
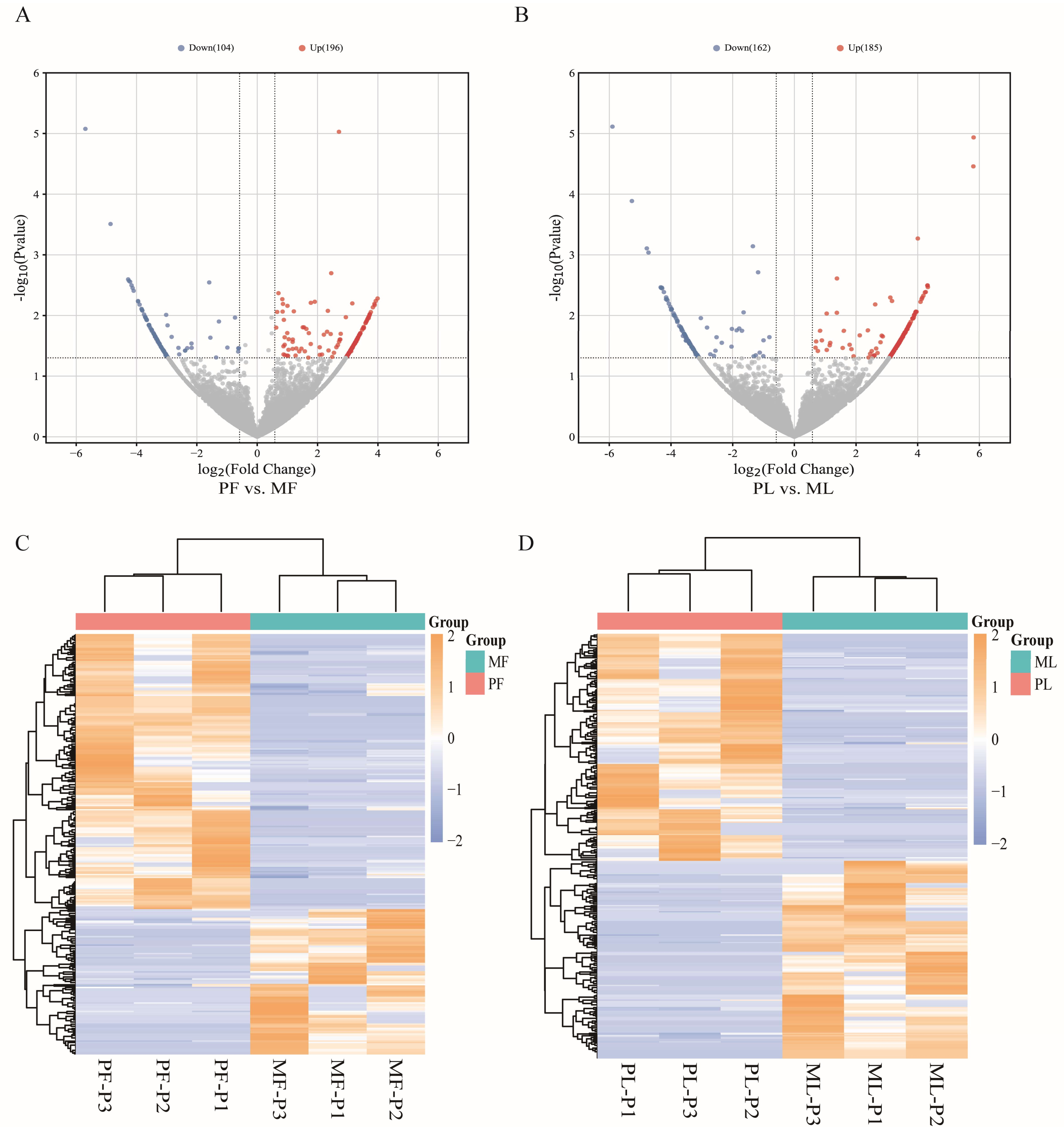
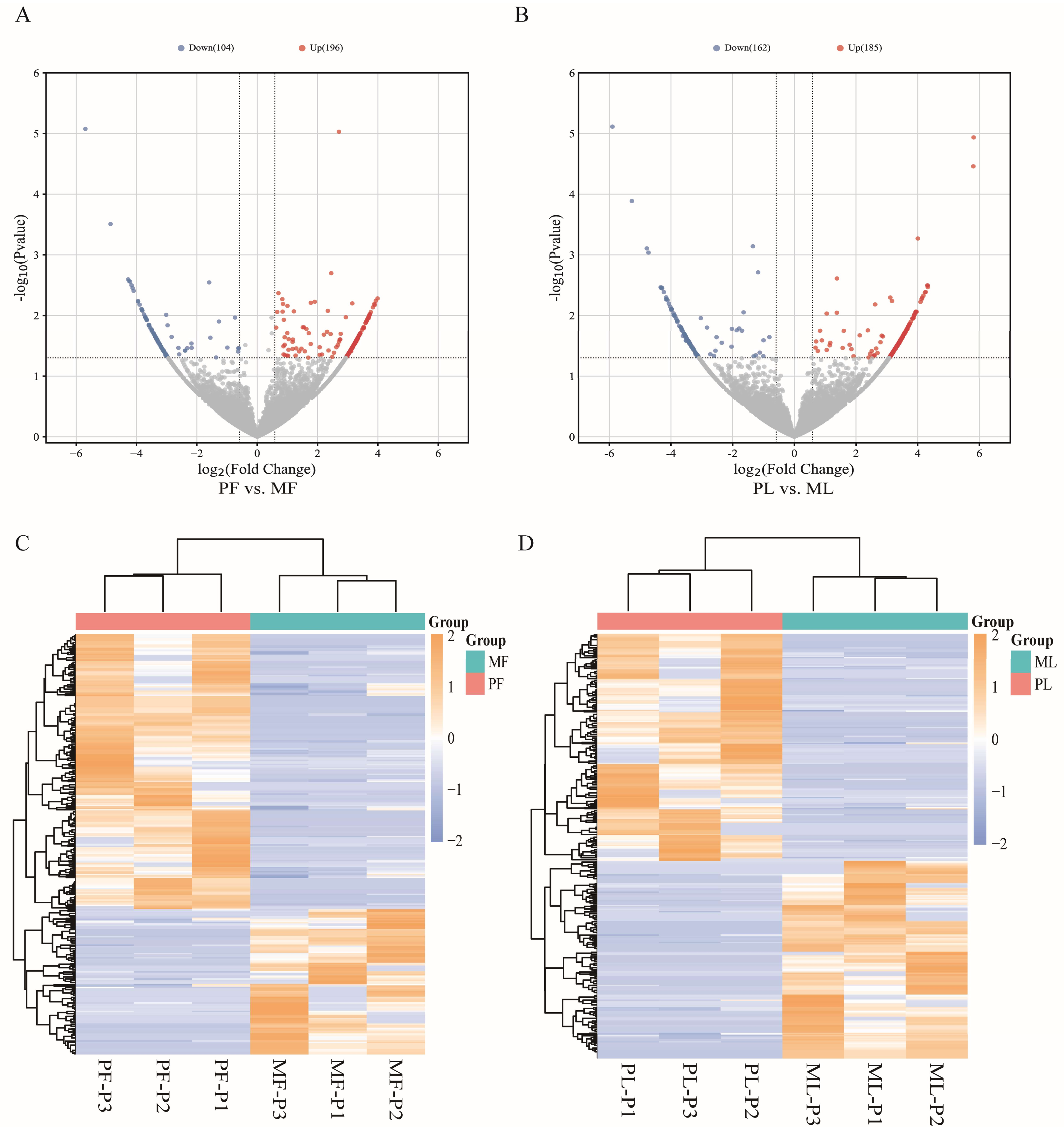
3.2. Identification of DE circRNAs
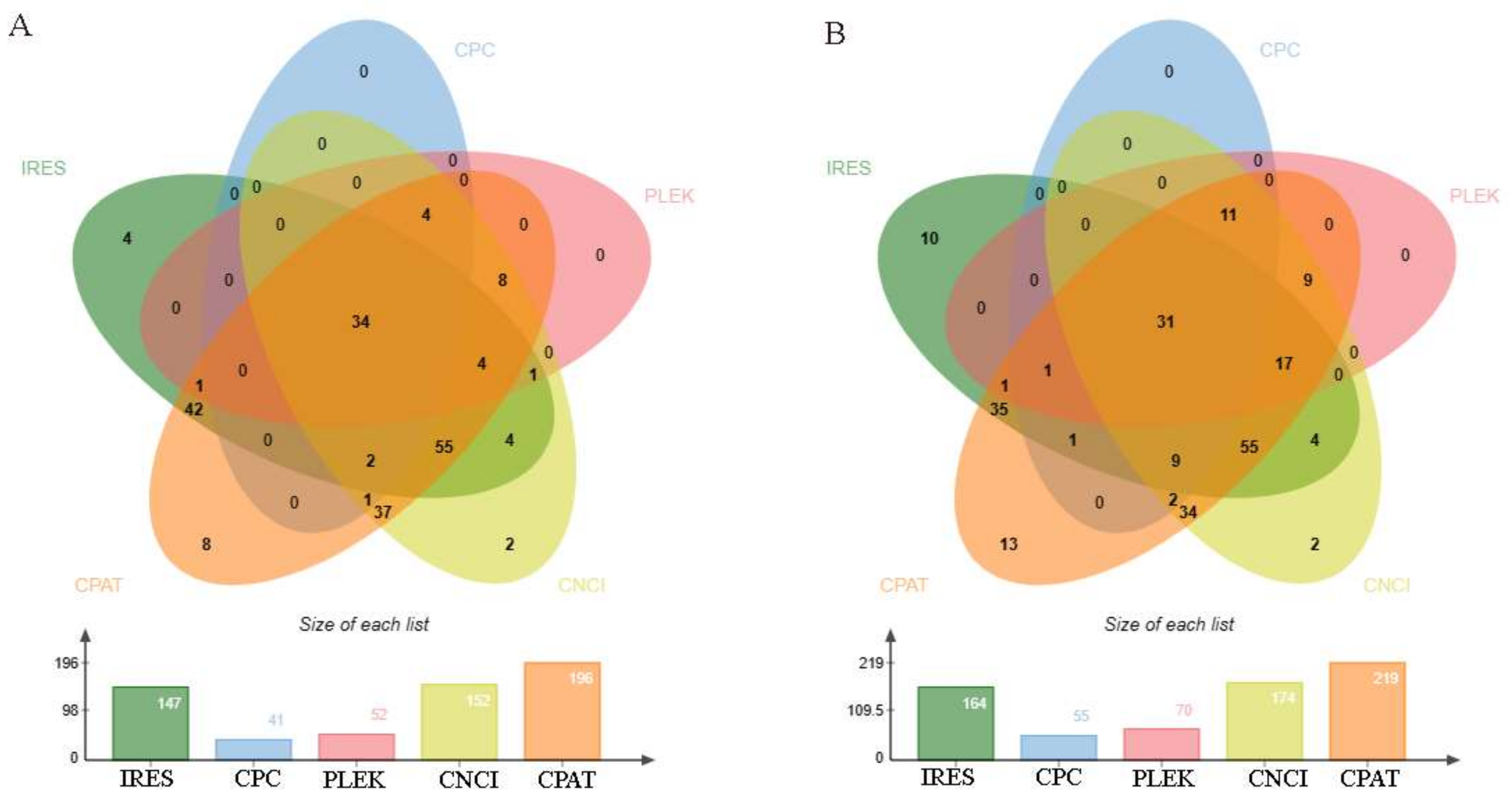
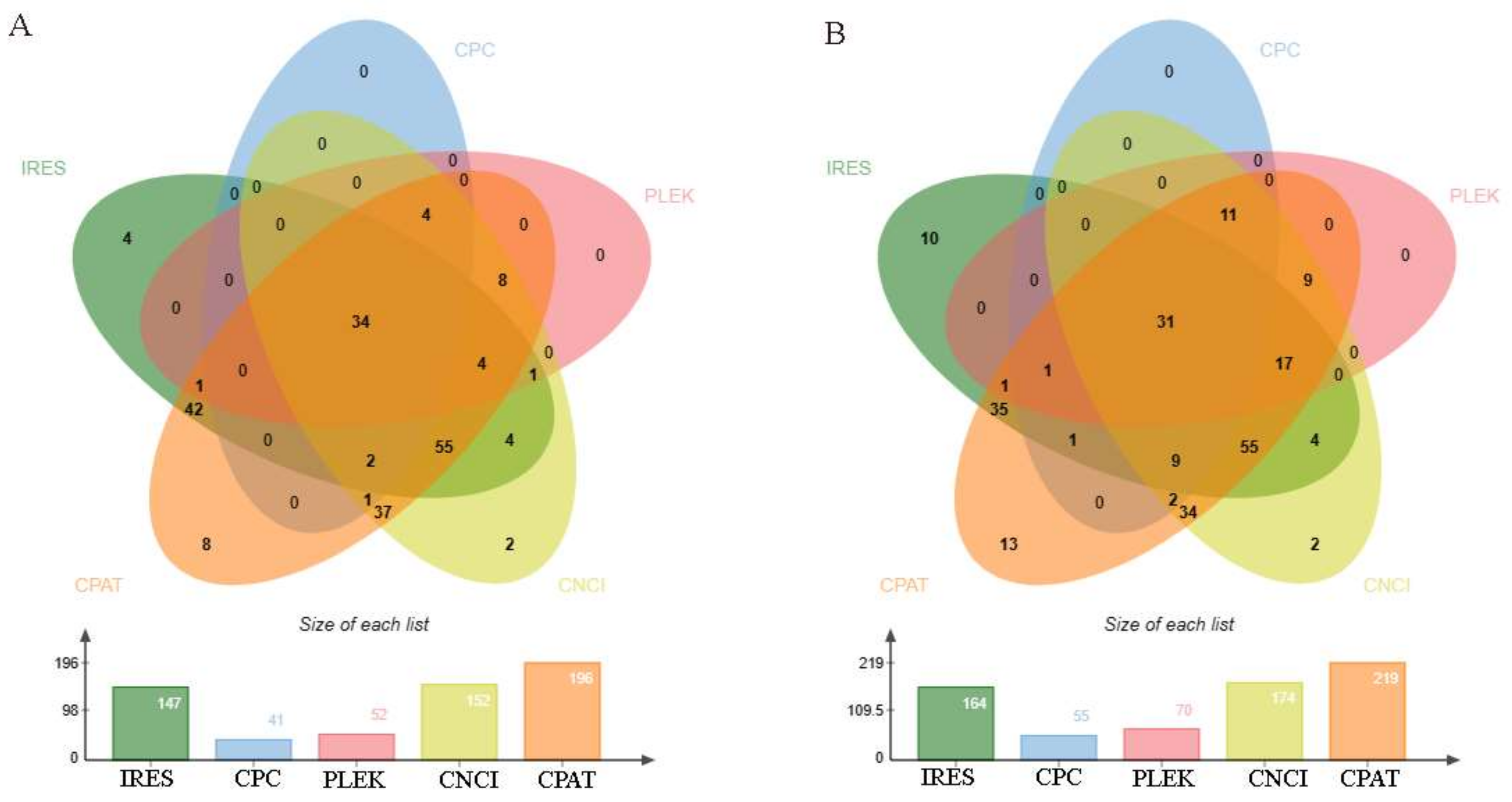
3.3. Identifying the Coding Potential of DE circRNAs
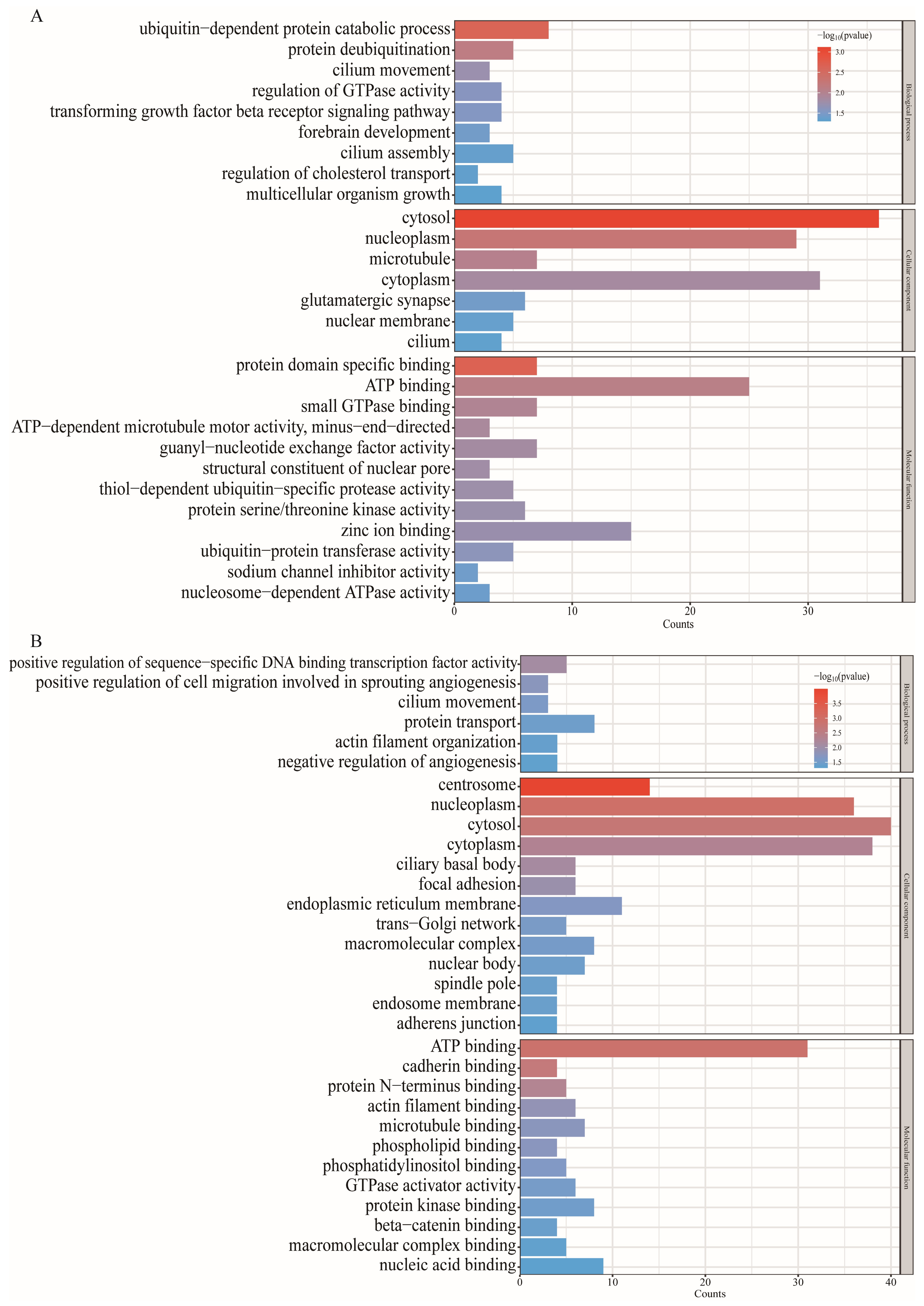
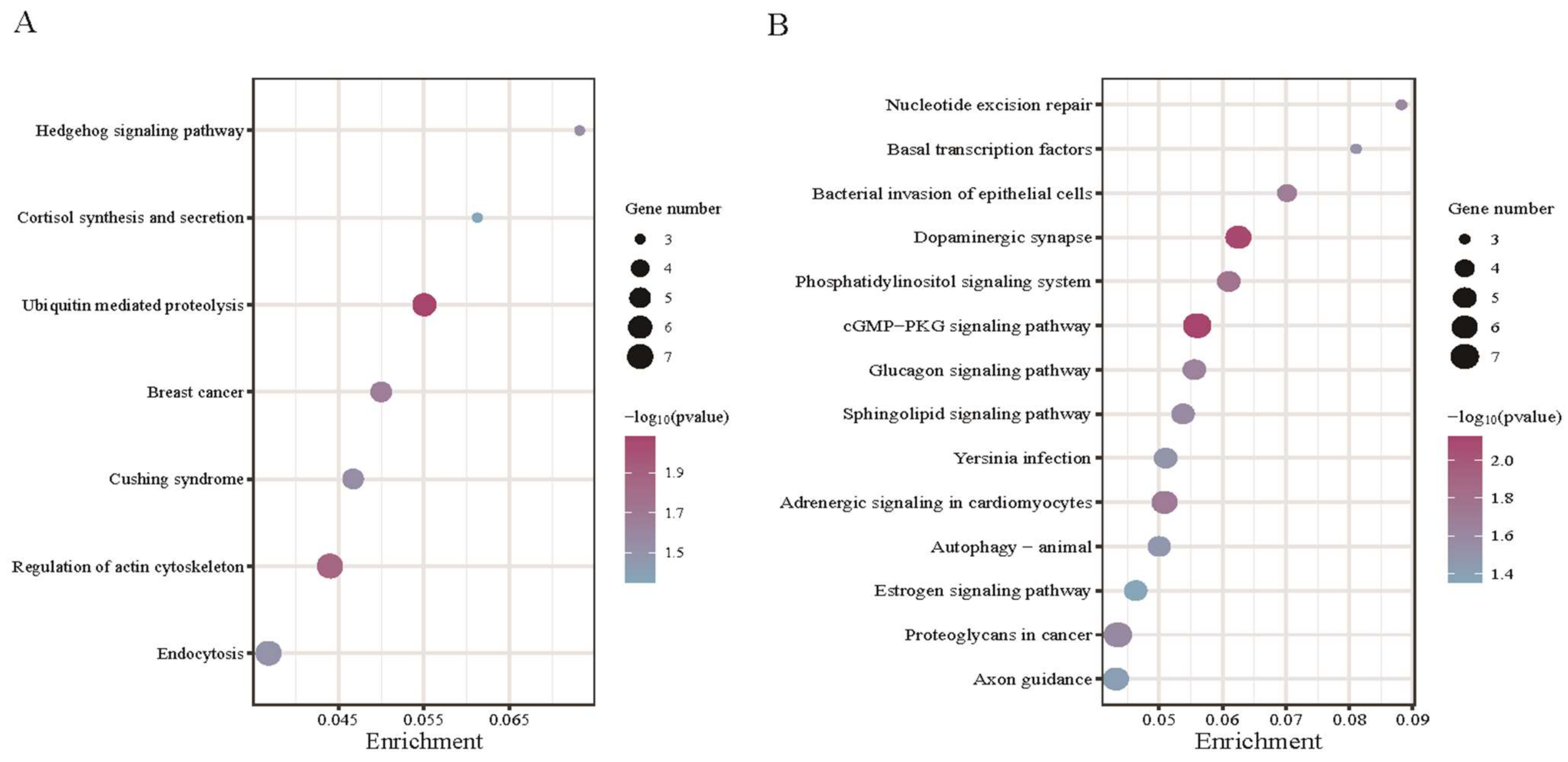
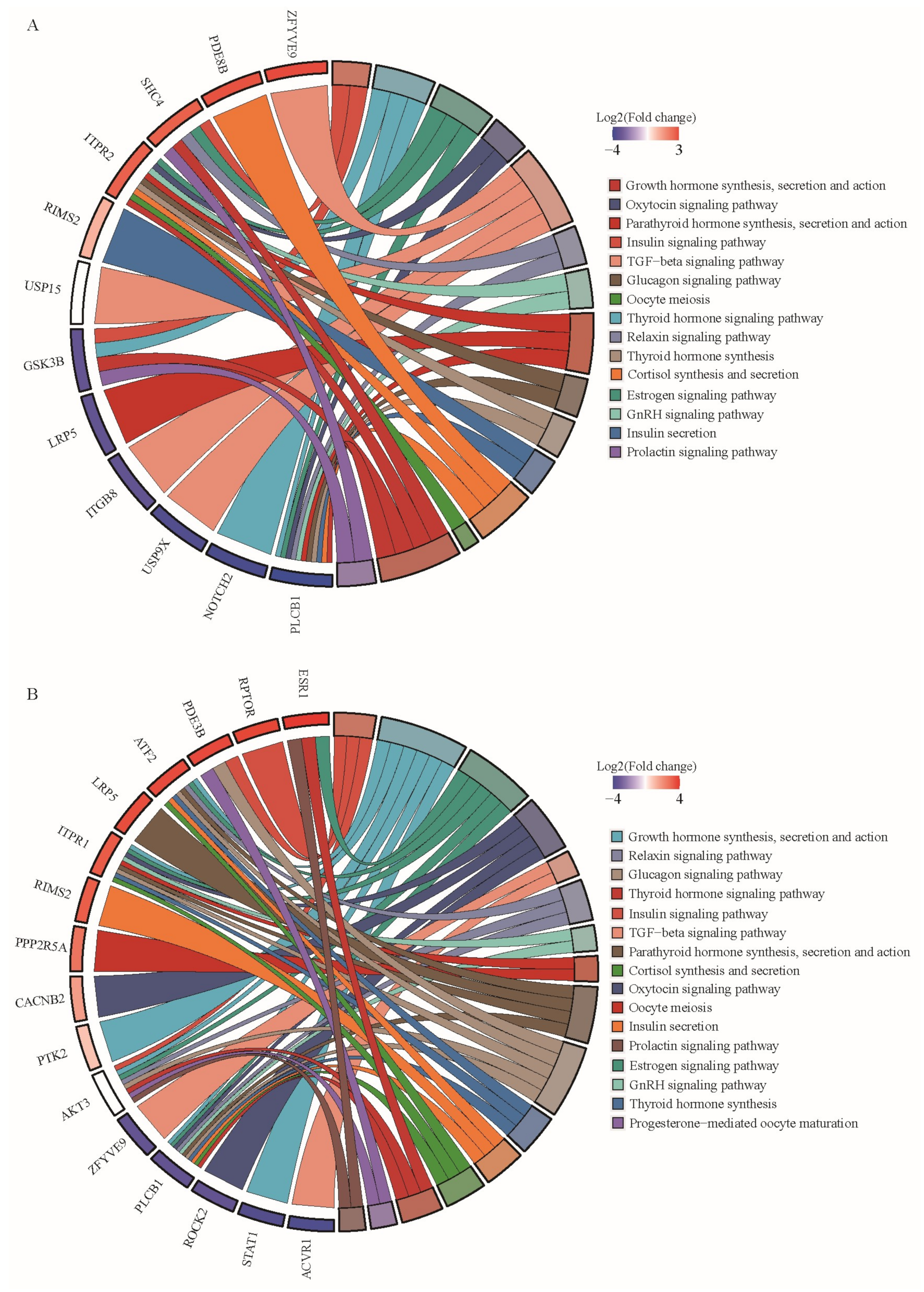
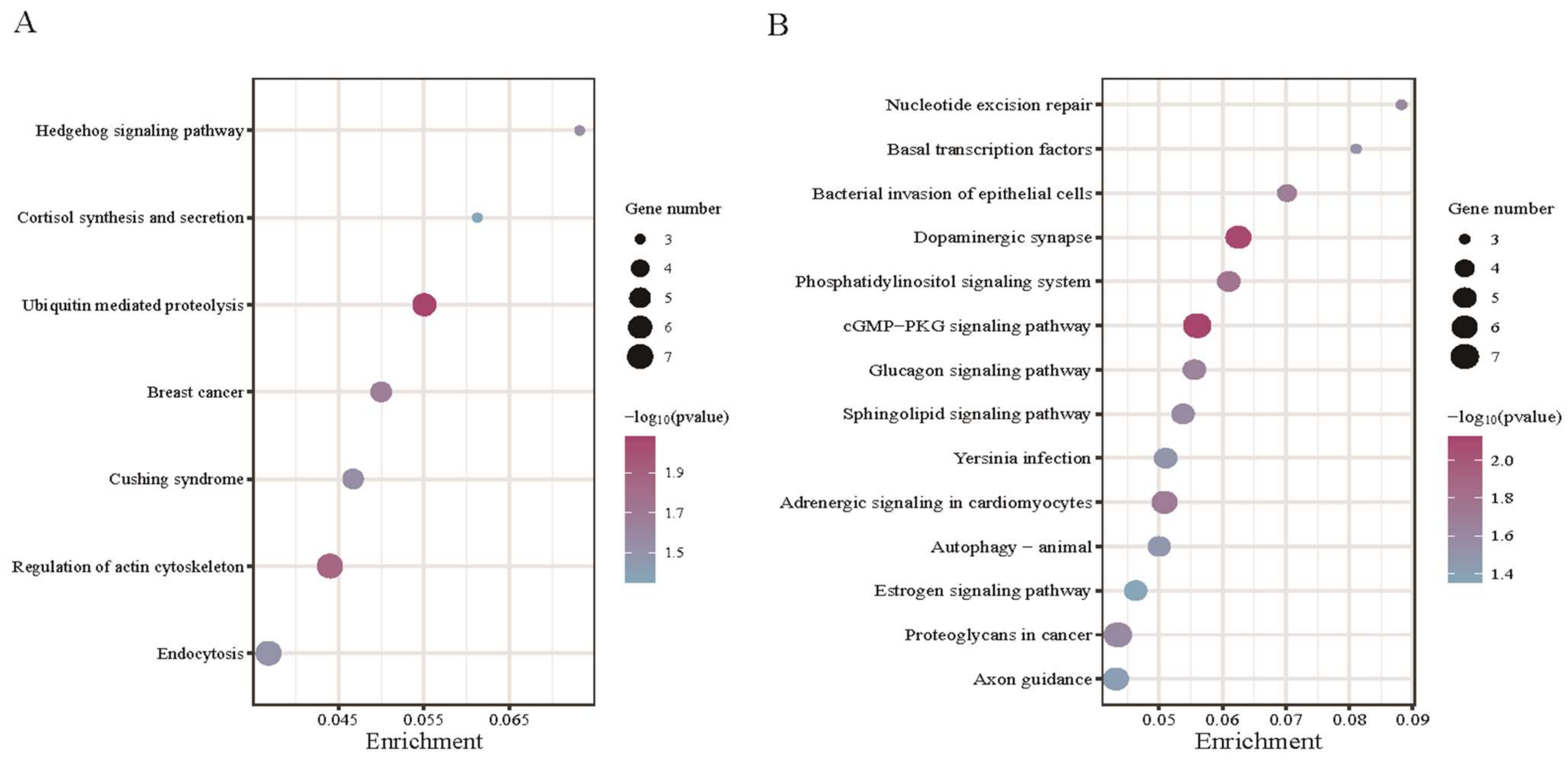
3.4. Functional Enrichment for Source Genes of DE circRNAs
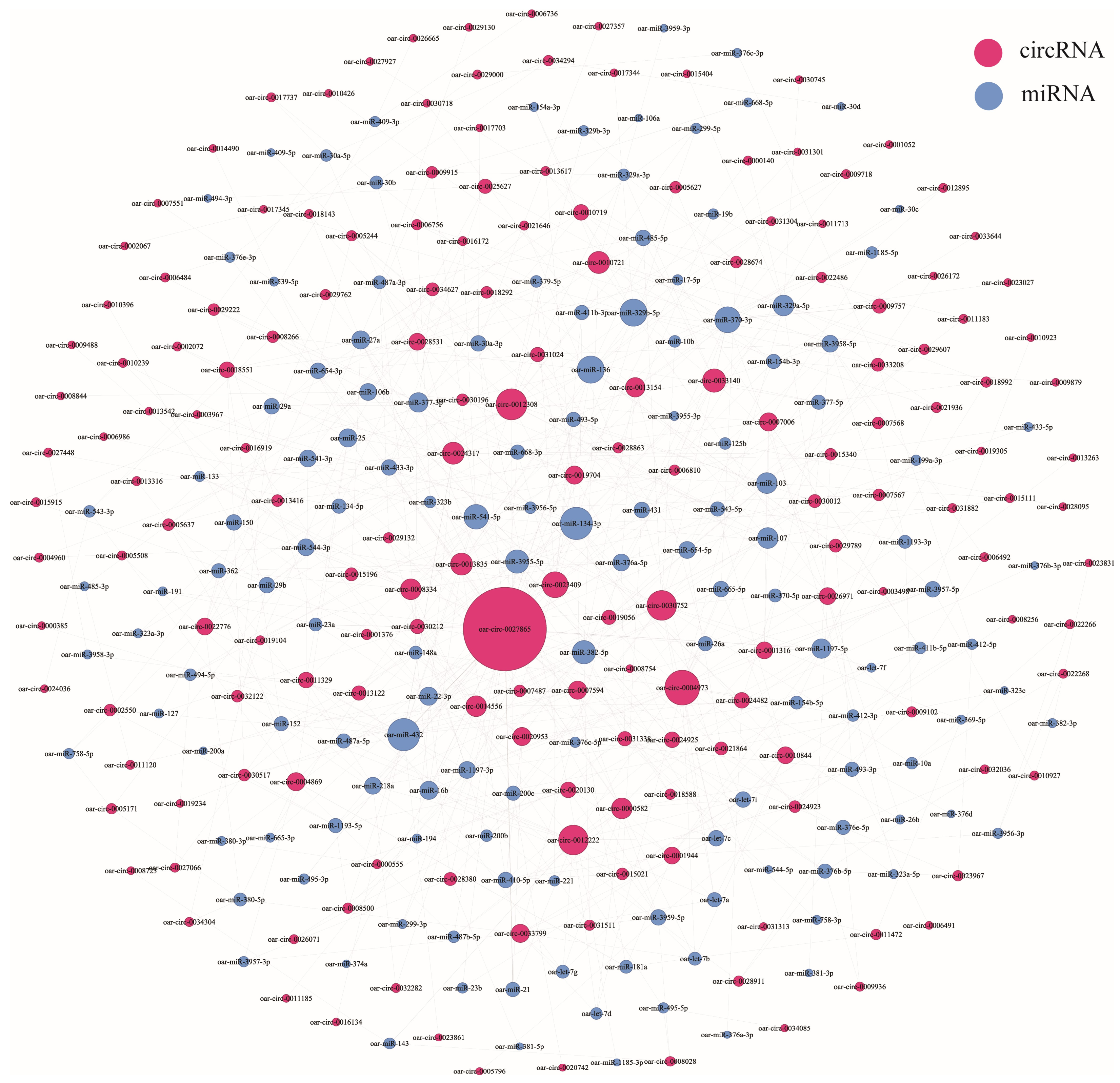
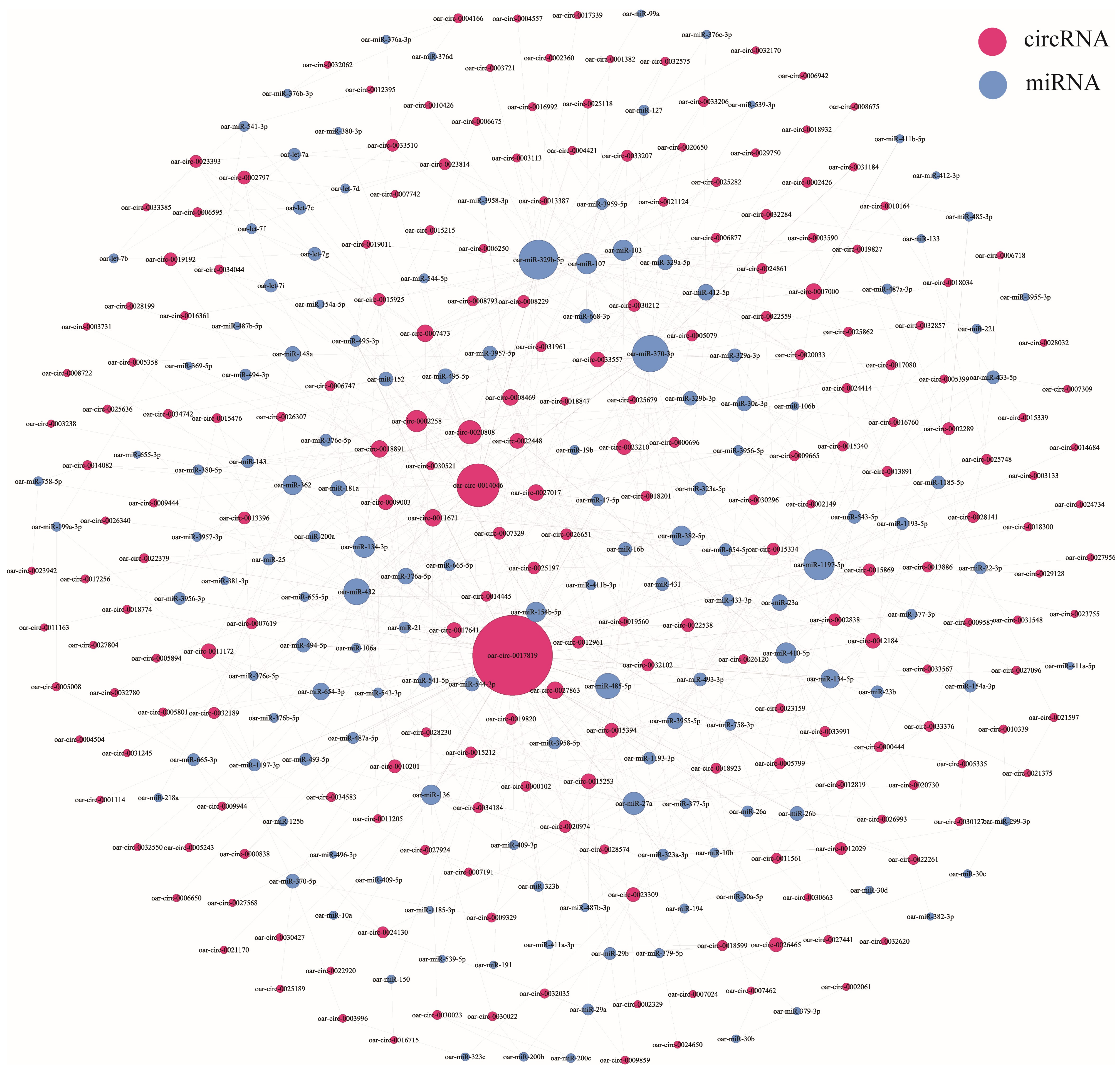
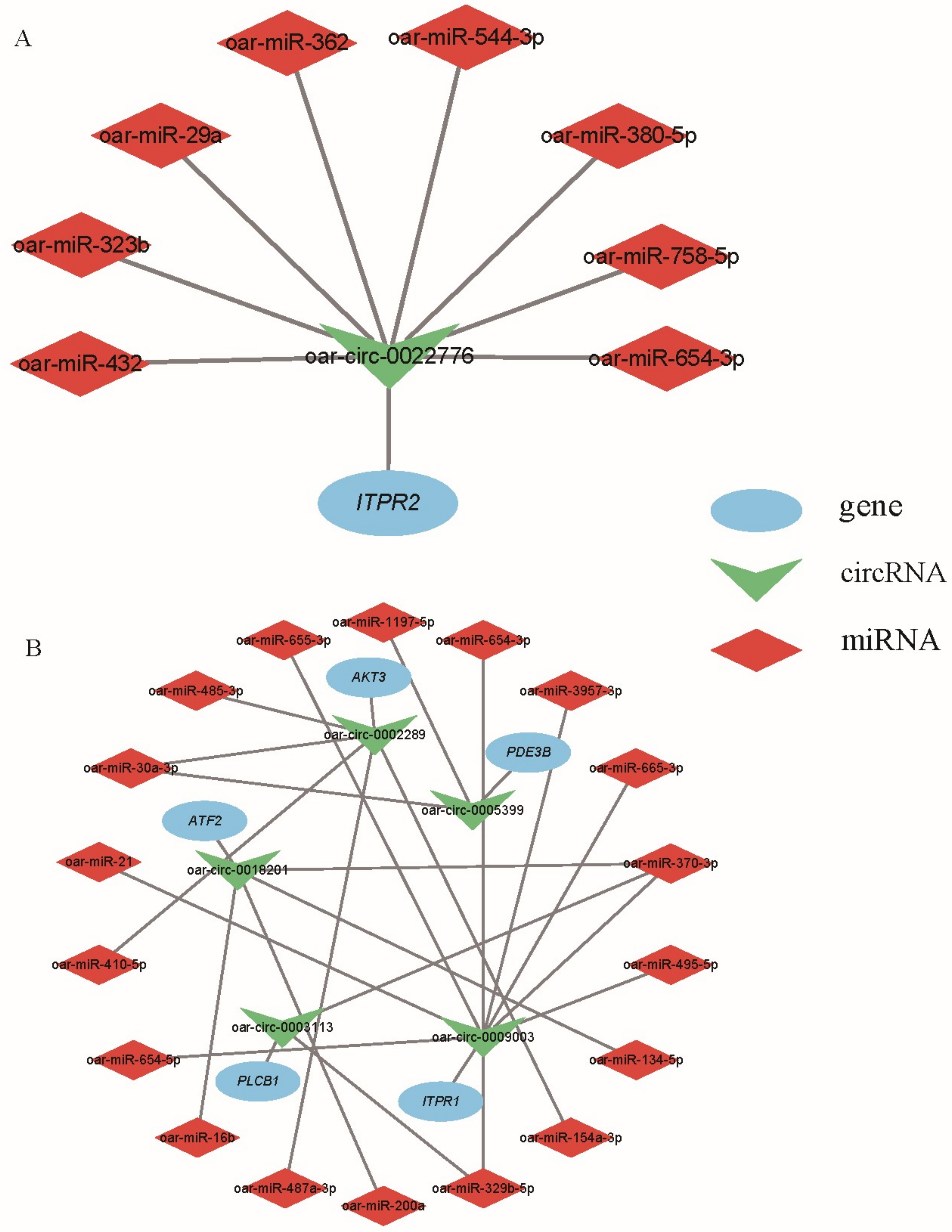
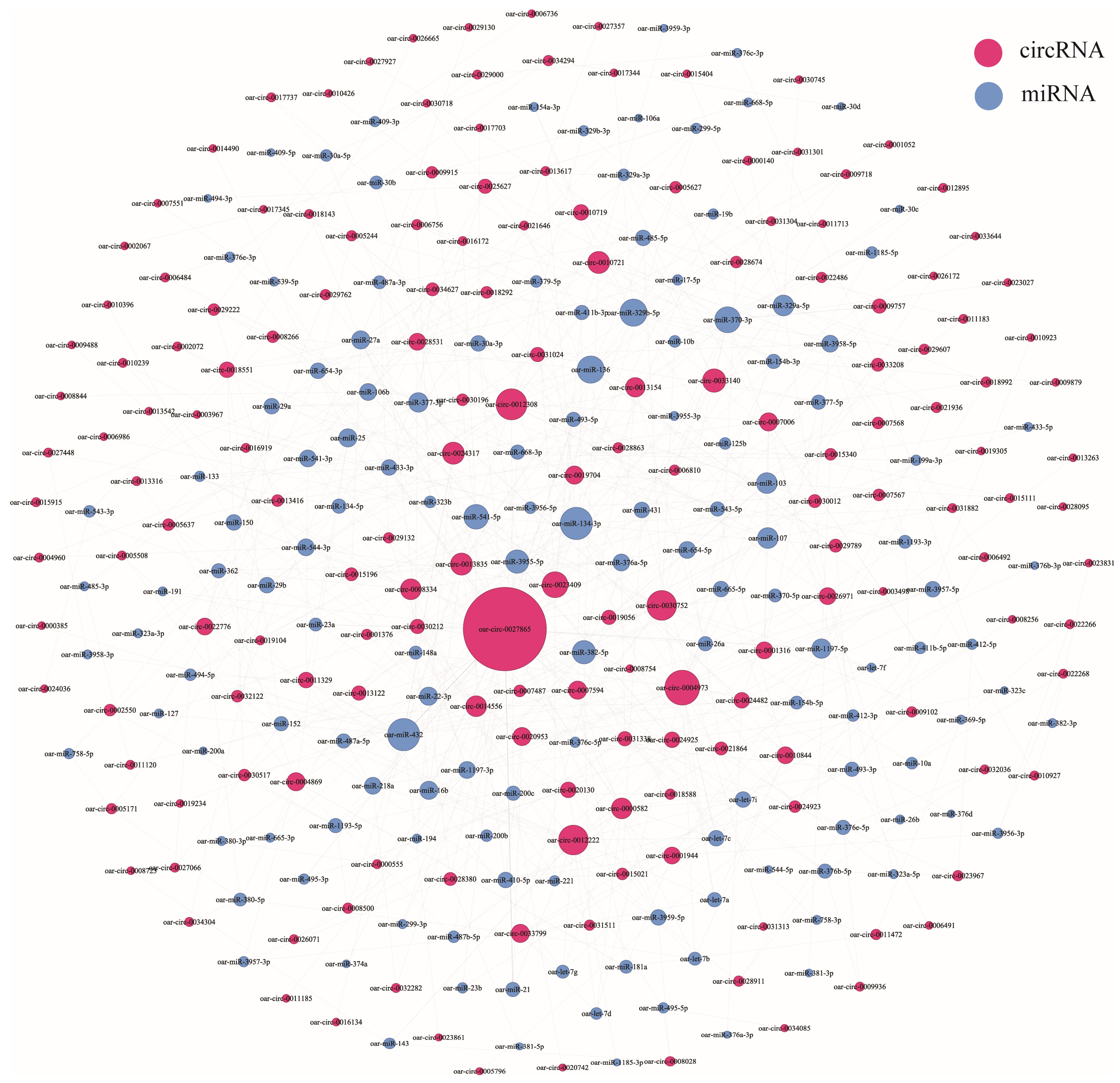
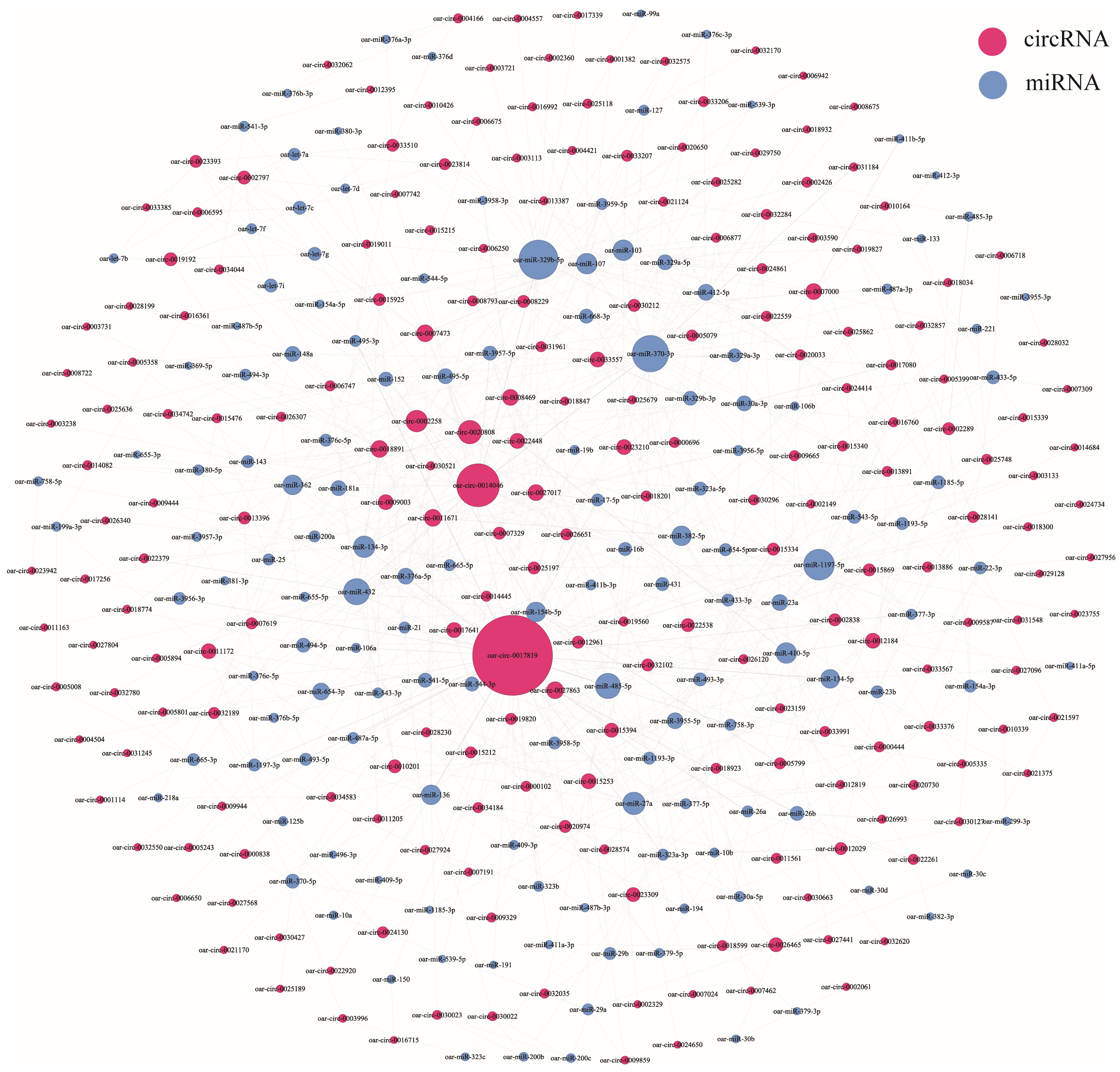
3.5. CircRNA–miRNA Coexpression Network
4. Discussion
4.1. Characteristics of Pituitary circRNAs
4.2. Functional Enrichment Analysis of DE circRNAs of Their Source Genes
4.3. Functional Exploration of Key circRNAs and Their Source Genes
4.4. The Clinical Significance of the Findings
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chu, M.X.; Liu, Z.H.; Jiao, C.L.; He, Y.Q.; Fang, L.; Ye, S.C.; Chen, G.H.; Wang, J.Y. Mutations in BMPR-IB and BMP-15 genes are associated with litter size in Small Tailed Han sheep (Ovis aries). J. Anim. Sci. 2007, 85, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Ding, Y.; Liu, J.; Liu, Y.; Zhao, X.; Li, G.; Zhang, C.; Li, C.; Wang, Y.; Kalds, P.; et al. Highly efficient generation of sheep with a defined FecB(B) mutation via adenine base editing. Genet. Sel. Evol. 2020, 52, 35. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, N.M. A review of the effects of the Booroola gene (FecB) on sheep production. Small Rumin. Res. 2009, 85, 75–84. [Google Scholar] [CrossRef]
- Davis, G.H.; Balakrishnan, L.; Ross, I.K.; Wilson, T.; Galloway, S.M.; Lumsden, B.M.; Hanrahan, J.P.; Mullen, M.; Mao, X.Z.; Wang, G.L.; et al. Investigation of the Booroola (FecB) and Inverdale (FecX(I)) mutations in 21 prolific breeds and strains of sheep sampled in 13 countries. Anim. Reprod. Sci. 2006, 92, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Notter, D.R. Genetic aspects of reproduction in sheep. Reprod. Domest. Anim. 2008, 43 (Suppl. S2), 122–128. [Google Scholar] [CrossRef]
- Hua, G.H.; Yang, L.G. A review of research progress of FecB gene in Chinese breeds of sheep. Anim. Reprod. Sci. 2009, 116, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.L.; Guo, X.F.; Ma, L.; Zhang, X.S.; Zhang, J.L.; Zhao, S.G.; Chu, M.X. The expression and mutation of BMPR1B and its association with litter size in small-tail Han sheep (Ovis aries). Arch. Anim. Breed. 2021, 64, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, X.; He, X.; Liu, Q.; Di, R.; Hu, W.; Cao, X.; Zhang, X.; Zhang, J.; Chu, M. Effects of FecB Mutation on Estrus, Ovulation, and Endocrine Characteristics in Small Tail Han Sheep. Front. Vet. Sci. 2021, 8, 709737. [Google Scholar] [CrossRef]
- Abreu, A.P.; Kaiser, U.B. Pubertal development and regulation. Lancet Diabetes Endocrinol. 2016, 4, 254–264. [Google Scholar] [CrossRef]
- Sang, Q.; Ray, P.F.; Wang, L. Understanding the genetics of human infertility. Science 2023, 380, 158–163. [Google Scholar] [CrossRef]
- Isaacs, K.L.; McNatty, K.P.; Condell, L.; Shaw, L.; Heath, D.A.; Hudson, N.L.; Littlejohn, R.P.; McLeod, B.J. Plasma FSH, LH and immunoreactive inhibin concentrations in FecBB/FecBB and FecB +/FecB + Booroola ewes and rams from birth to 12 months of age. J. Reprod. Fertil. 1995, 103, 89–97. [Google Scholar] [CrossRef] [PubMed]
- McNatty, K.P.; Hudson, N.L.; Lun, S.; Heath, D.A.; Shaw, L.; Condell, L.; Phillips, D.J.; Clarke, I.J. Gonadotrophin-releasing hormone and the control of ovulation rate by the FecB gene in Booroola ewes. J. Reprod. Fertil. 1993, 98, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.J.; Hudson, N.L.; McNatty, K.P. Effects of ovariectomy and genotype on bioactive FSH in plasma and pituitary of Booroola ewes. J. Reprod. Fertil. 1993, 98, 559–565. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F. Oxidative DNA damage and repair at non-coding regulatory regions. Trends Cell Biol. 2023, 23, S0962. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Ding, Y.; Kong, X.; Feng, G.; Xiang, W.; Chen, L.; Yang, F.; Zhang, K.; Chu, M.; Wang, P.; et al. Reproductive role of miRNA in the hypothalamic-pituitary axis. Mol. Cell Neurosci. 2018, 88, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, Z.; Yang, H.; Yao, X.; Yang, P.; Ren, C.; Wang, F.; Zhang, Y. Pituitary Transcriptomic Study Reveals the Differential Regulation of lncRNAs and mRNAs Related to Prolificacy in Different FecB Genotyping Sheep. Genes 2019, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, X.; Ma, Q.; Zhang, X.; Cao, Y.; Yao, Y.; You, S.; Wang, D.; Quan, R.; Hou, X.; et al. Genome-wide analysis of circular RNAs in prenatal and postnatal pituitary glands of sheep. Sci. Rep. 2017, 7, 16143. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Guo, Y.; Wei, X.; Peng, Y. Structure-Mediated Degradation of CircRNAs. Trends Cell Biol. 2020, 30, 501–503. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Sun, Z.; Hong, Q.; Liu, Y.; He, X.; Di, R.; Wang, X.; Ren, C.; Zhang, Z.; Chu, M. Characterization of circular RNA profiles of oviduct reveal the potential mechanism in prolificacy trait of goat in the estrus cycle. Front. Physiol. 2022, 13, 990691. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Shan, G. What happens at or after transcription: Insights into circRNA biogenesis and function. Transcription 2015, 6, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chen, K.; Chen, W.; Wang, J.; Chang, L.; Deng, J.; Wei, L.; Han, L.; Huang, C.; He, C. circRIP: An accurate tool for identifying circRNA-RBP interactions. Brief. Bioinform. 2022, 23, bbac186. [Google Scholar] [CrossRef]
- He, L.; Man, C.; Xiang, S.; Yao, L.; Wang, X.; Fan, Y. Circular RNAs’ cap-independent translation protein and its roles in carcinomas. Mol. Cancer 2021, 20, 119. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Wei, J.; Ni, W.; Xu, Y.; Yao, R.; Zhang, M.; Li, H.; Liu, L.; Dang, H.; et al. Comprehensive Expression Profiling Analysis of Pituitary Indicates that circRNA Participates in the Regulation of Sheep Estrus. Genes 2019, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Bartlewski, P.M.; Baby, T.E.; Giffin, J.L. Reproductive cycles in sheep. Anim. Reprod. Sci. 2011, 124, 259–268. [Google Scholar] [CrossRef]
- Wan, Z.; Yang, H.; Cai, Y.; Ma, J.; Cheng, P.; Wang, Z.; Wang, F.; Zhang, Y. Comparative Transcriptomic Analysis of Hu Sheep Pituitary Gland Prolificacy at the Follicular and Luteal Phases. Genes 2022, 13, 440. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chen, R.; Wang, S.K.; Belk, J.A.; Amaya, L.; Li, Z.; Cardenas, A.; Abe, B.T.; Chen, C.K.; Wender, P.A.; Chang, H.Y. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 2023, 41, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wu, J.; Xu, T.; Yang, Q.; He, J.; Song, X. IRESfinder: Identifying RNA internal ribosome entry site in eukaryotic cell using framed k-mer features. J. Genet. Genomics 2018, 45, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wei, S.; Kang, X.; Yang, C.; Wang, F.; Dai, T.; Guo, X.; Ma, Z.; Li, C.; Zhao, H.; Dan, X. Analysis of reproduction-related transcriptomes on pineal-hypothalamic-pituitary-ovarian tissues during estrus and anestrus in Tan sheep. Front. Vet. Sci. 2022, 9, 1068882. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; He, X.; Zhu, M.; Gan, S.; Guo, X.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Comparative Transcriptomics Identify Key Hypothalamic Circular RNAs that Participate in Sheep (Ovis aries) Reproduction. Animals 2019, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- La, Y.; Tang, J.; Di, R.; Wang, X.; Liu, Q.; Zhang, L.; Zhang, X.; Zhang, J.; Hu, W.; Chu, M. Differential Expression of Circular RNAs in Polytocous and Monotocous Uterus during the Reproductive Cycle of Sheep. Animals 2019, 9, 797. [Google Scholar] [CrossRef] [PubMed]
- Gebreselassie, G.; Berihulay, H.; Jiang, L.; Ma, Y. Review on Genomic Regions and Candidate Genes Associated with Economically Important Production and Reproduction Traits in Sheep (Ovies aries). Animals 2019, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, C.; Wang, L.; Feng, R.; Guo, Y.; Feng, S.; Zhang, L.; Zheng, Z.; Su, G.; Fan, L.; et al. Correlation analysis of serum reproductive hormones and metabolites during multiple ovulation in sheep. BMC Vet. Res. 2022, 18, 290. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.N.; Stackpole, C.A.; Breen, K.M.; Clarke, I.J.; Karsch, F.J.; Rivalland, E.T.; Turner, A.I.; Caddy, D.J.; Wagenmaker, E.R.; Oakley, A.E.; et al. Estradiol enables cortisol to act directly upon the pituitary to suppress pituitary responsiveness to GnRH in sheep. Neuroendocrinology 2009, 89, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.T.; Goodman, R.L.; Hileman, S.M.; Lehman, M.N. Evidence that synaptic plasticity of glutamatergic inputs onto KNDy neurones during the ovine follicular phase is dependent on increasing levels of oestradiol. J. Neuroendocrinol. 2021, 33, e12945. [Google Scholar] [CrossRef]
- Mangla, A.; Guerra, M.T.; Nathanson, M.H. Type 3 inositol 1,4,5-trisphosphate receptor: A calcium channel for all seasons. Cell Calcium. 2020, 85, 102132. [Google Scholar] [CrossRef]
- Tarsani, E.; Kranis, A.; Maniatis, G.; Avendano, S.; Hager-Theodorides, A.L.; Kominakis, A. Deciphering the mode of action and position of genetic variants impacting on egg number in broiler breeders. BMC Genom. 2020, 21, 512. [Google Scholar] [CrossRef]
- Sun, B.; Ni, M.; Tian, S.; Guo, W.; Cai, S.; Sondergaard, M.T.; Chen, Y.; Mu, Y.; Estillore, J.P.; Wang, R.; et al. A gain-of-function mutation in the ITPR1 gating domain causes male infertility in mice. J. Cell Physiol. 2022, 237, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.S.; Marco-Jiménez, F.; Pérez-García, M.; Naturil-Alfonso, C.; Peñaranda, D.S.; Viudes-de-Castro, M.P. Oocyte quality and in vivo embryo survival after ovarian stimulation in nulliparous and multiparous rabbit does. Theriogenology 2022, 189, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.; Mishra, B.; Bhaskar, R.; Vikas, Y.N.V.; Umesh, A.; Guttula, P.K.; Gupta, M.K. Analyzing the effect of heparin on in vitro capacitation and spermatozoal RNA population in goats. Int. J. Biol. Macromol. 2023, 241, 124502. [Google Scholar] [CrossRef]
- Yi, S.; Liu, L.F.; Zhou, L.F.; Zhao, B.W.; Wang, W.M.; Gao, Z.X. Screening of Biomarkers Related to Ovarian Maturation and Spawning in Blunt Snout Bream (Megalobrama amblycephala) Based on Metabolomics and Transcriptomics. Mar. Biotechnol. 2020, 22, 180–193. [Google Scholar] [CrossRef]
- Gholizadeh, M.; Esmaeili-Fard, S.M. Meta-analysis of genome-wide association studies for litter size in sheep. Theriogenology 2022, 180, 103–112. [Google Scholar] [CrossRef]
- He, X.; Di, R.; Guo, X.; Cao, X.; Zhou, M.; Li, X.; Xia, Q.; Wang, X.; Zhang, J.; Zhang, X.; et al. Transcriptomic Changes of Photoperiodic Response in the Hypothalamus Were Identified in Ovariectomized and Estradiol-Treated Sheep. Front. Mol. Biosci. 2022, 9, 848144. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Chen, X.; Liu, M.; Zhang, L.; Ma, X.; Tian, S. Differential Expression and Functional Analysis of CircRNA in the Ovaries of Low and High Fecundity Hanper Sheep. Animals 2021, 11, 1863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, J.; Di, R.; Liu, Q.; Wang, X.; Gan, S.; Zhang, X.; Zhang, J.; Chu, M.; Hu, W. Integrated Hypothalamic Transcriptome Profiling Reveals the Reproductive Roles of mRNAs and miRNAs in Sheep. Front. Genet. 2019, 10, 1296. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Chu, M. miRNA-mRNA analysis of sheep adrenal glands reveals the network regulating reproduction. BMC Genom. Data 2022, 23, 44. [Google Scholar] [CrossRef]
- Wan, Z.; Yang, H.; Chen, P.; Wang, Z.; Cai, Y.; Yao, X.; Wang, F.; Zhang, Y. The Novel Competing Endogenous Long Noncoding RNA SM2 Regulates Gonadotropin Secretion in the Hu Sheep Anterior Pituitary by Targeting the Oar-miR-16b/TGF-β/SMAD2 Signaling Pathway. Cells 2022, 11, 985. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Tang, J.; He, X.; Di, R.; Zhang, X.; Zhang, J.; Guo, X.; Chu, M.; Hu, W. Comparative Transcriptomics Identify Key Pituitary Circular RNAs That Participate in Sheep (Ovis aries) Reproduction. Animals 2023, 13, 2711. https://doi.org/10.3390/ani13172711
Yang J, Tang J, He X, Di R, Zhang X, Zhang J, Guo X, Chu M, Hu W. Comparative Transcriptomics Identify Key Pituitary Circular RNAs That Participate in Sheep (Ovis aries) Reproduction. Animals. 2023; 13(17):2711. https://doi.org/10.3390/ani13172711
Chicago/Turabian StyleYang, Jianqi, Jishun Tang, Xiaoyun He, Ran Di, Xiaosheng Zhang, Jinlong Zhang, Xiaofei Guo, Mingxing Chu, and Wenping Hu. 2023. "Comparative Transcriptomics Identify Key Pituitary Circular RNAs That Participate in Sheep (Ovis aries) Reproduction" Animals 13, no. 17: 2711. https://doi.org/10.3390/ani13172711
APA StyleYang, J., Tang, J., He, X., Di, R., Zhang, X., Zhang, J., Guo, X., Chu, M., & Hu, W. (2023). Comparative Transcriptomics Identify Key Pituitary Circular RNAs That Participate in Sheep (Ovis aries) Reproduction. Animals, 13(17), 2711. https://doi.org/10.3390/ani13172711