Correlation between Targeted qPCR Assays and Untargeted DNA Shotgun Metagenomic Sequencing for Assessing the Fecal Microbiota in Dogs

 ,
,  , , , , , , , , , ,
, , , , , , , , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Quantitative PCR and Dysbiosis Index (DI)
2.3. Shotgun Metagenomic Sequencing
2.4. Statistical Analyses
3. Results
3.1. Dysbiosis Index of the Study Population
3.2. Alpha Diversity of the Fecal Microbiota in the Study Population
3.3. Beta Diversity of the Fecal Microbiota in the Study Population
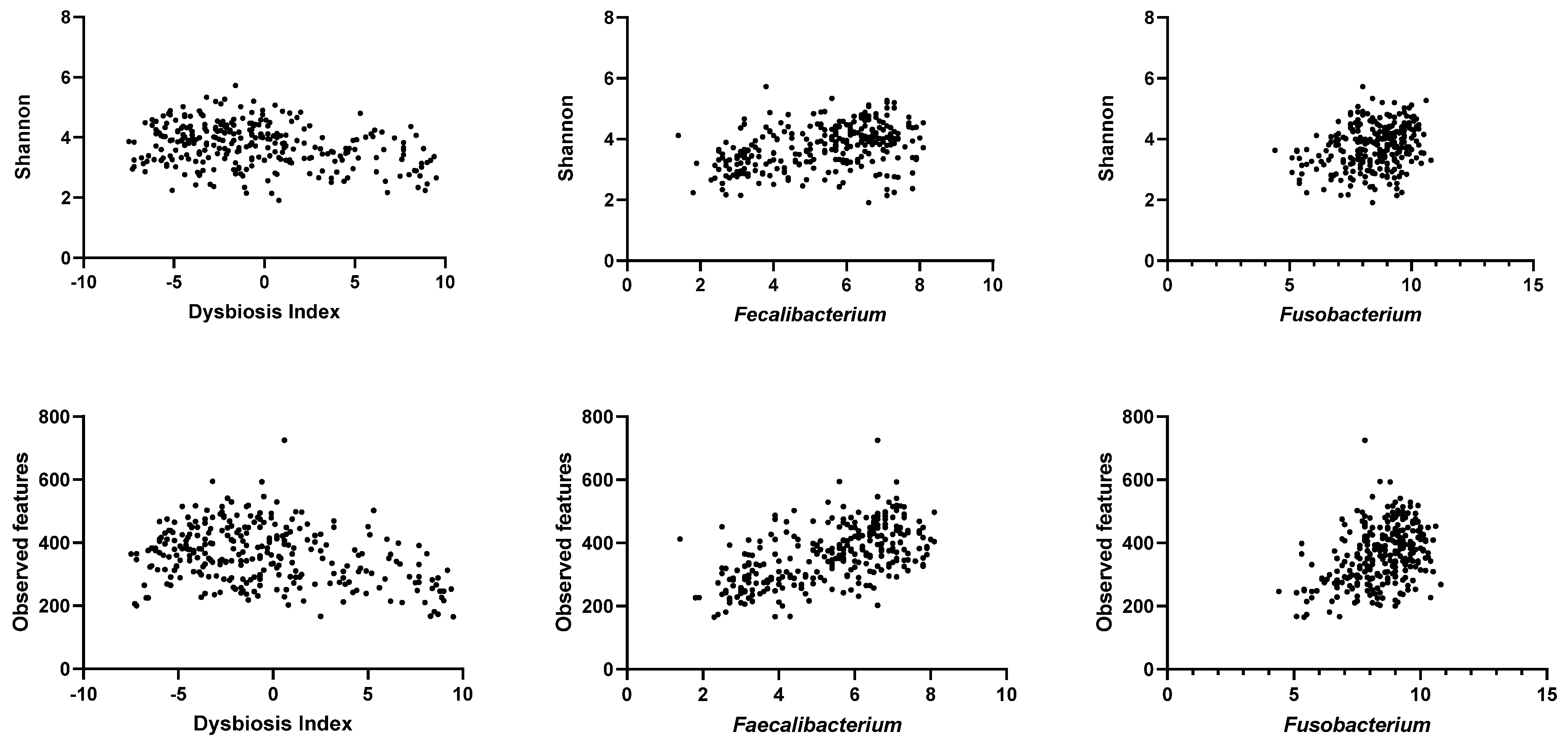
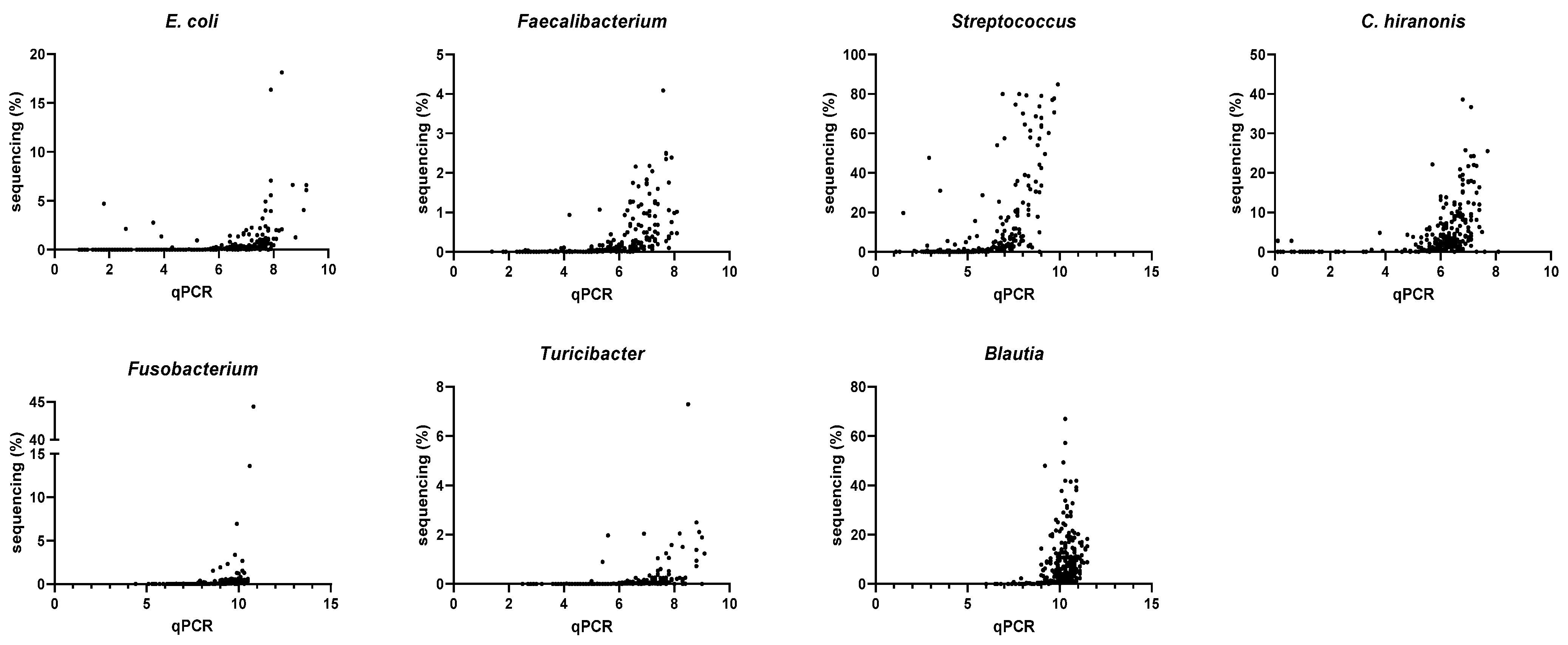
3.4. Correlation between qPCR-Based Dysbiosis Index and Shotgun Metagenomic Sequencing Data
3.5. Core Microbiota in Healthy Dogs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bai, H.; Liu, T.; Wang, S.; Shen, L.; Wang, Z. Variations in gut microbiome and metabolites of dogs with acute diarrhea in poodles and Labrador retrievers. Arch. Microbiol. 2023, 205, 97. [Google Scholar] [CrossRef] [PubMed]
- Herstad, K.M.V.; Trosvik, P.; Haaland, A.H.; Haverkamp, T.H.A.; de Muinck, E.J.; Skancke, E. Changes in the fecal microbiota in dogs with acute hemorrhagic diarrhea during an outbreak in Norway. J. Vet. Intern. Med. 2021, 35, 2177–2186. [Google Scholar] [CrossRef]
- Minamoto, Y.; Otoni, C.C.; Steelman, S.M.; Büyükleblebici, O.; Steiner, J.M.; Jergens, A.E.; Suchodolski, J.S. Alteration of the fecal microbiota and serum metabolite profiles in dogs with idiopathic inflammatory bowel disease. Gut Microbes 2015, 6, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Xenoulis, P.G.; Palculict, B.; Allenspach, K.; Steiner, J.M.; Van House, A.M.; Suchodolski, J.S. Molecular-phylogenetic characterization of microbial communities imbalances in the small intestine of dogs with inflammatory bowel disease. FEMS Microbiol. Ecol. 2008, 66, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Bharti, R.; Grimm, D.G. Current challenges and best-practice protocols for microbiome analysis. Brief. Bioinform. 2021, 22, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L.; et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [PubMed]
- Atherly, T.; Rossi, G.; White, R.; Seo, Y.J.; Wang, C.; Ackermann, M.; Breuer, M.; Allenspach, K.; Mochel, J.P.; Jergens, A.E. Glucocorticoid and dietary effects on mucosal microbiota in canine inflammatory bowel disease. PLoS ONE 2019, 14, e0226780. [Google Scholar] [CrossRef]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef]
- Engelbrektson, A.; Kunin, V.; Wrighton, K.C.; Zvenigorodsky, N.; Chen, F.; Ochman, H.; Hugenholtz, P. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010, 4, 642–647. [Google Scholar] [CrossRef]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.; Nash, A.; Li, Q.; Ahn, T.-H. Comparison of 16S and whole genome dog microbiomes using machine learning. BioData Min. 2021, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Tanprasertsuk, J.; Jha, A.R.; Shmalberg, J.; Jones, R.B.; Perry, L.M.; Maughan, H.; Honaker, R.W. The microbiota of healthy dogs demonstrates individualized responses to synbiotic supplementation in a randomized controlled trial. Anim. Microbiome 2021, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- AlShawaqfeh, M.K.; Wajid, B.; Minamoto, Y.; Markel, M.; Lidbury, J.A.; Steiner, J.M.; Serpedin, E.; Suchodolski, J.S. A dysbiosis index to assess microbial changes in fecal samples of dogs with chronic inflammatory enteropathy. FEMS Microbiol. Ecol. 2017, 93, fix136. [Google Scholar] [CrossRef] [PubMed]
- Felix, A.P.; Souza, C.M.M.; de Oliveira, S.G. Biomarkers of gastrointestinal functionality in dogs: A systematic review and meta-analysis. Anim. Feed Sci. Technol. 2022, 283, 115183. [Google Scholar] [CrossRef]
- Werner, M.; Suchodolski, J.S.; Straubinger, R.K.; Wolf, G.; Steiner, J.M.; Lidbury, J.A.; Neuerer, F.; Hartmann, K.; Unterer, S. Effect of amoxicillin-clavulanic acid on clinical scores, intestinal microbiome, and amoxicillin-resistant Escherichia coli in dogs with uncomplicated acute diarrhea. J. Vet. Intern. Med. 2020, 34, 1166–1176. [Google Scholar] [CrossRef]
- Toresson, L.; Spillmann, T.; Pilla, R.; Ludvigsson, U.; Hellgren, J.; Olmedal, G.; Suchodolski, J.S. Clinical effects of faecal microbiota transplantation as adjunctive therapy in dogs with chronic enteropathies—A retrospective case series of 41 Dogs. Vet. Sci. 2023, 10, 271. [Google Scholar] [CrossRef]
- Pilla, R.; Gaschen, F.P.; Barr, J.W.; Olson, E.; Honneffer, J.; Guard, B.C.; Blake, A.B.; Villanueva, D.; Khattab, M.R.; AlShawaqfeh, M.K.; et al. Effects of metronidazole on the fecal microbiome and metabolome in healthy dogs. J. Vet. Intern. Med. 2020, 34, 1853–1866. [Google Scholar] [CrossRef]
- Belchik, S.E.; Oba, P.M.; Wyss, R.; Asare, P.T.; Vidal, S.; Miao, Y.; Adesokan, Y.; Suchodolski, J.S.; Swanson, K.S. Effects of a milk oligosaccharide biosimilar on fecal characteristics, microbiota, and bile acid, calprotectin, and immunoglobulin concentrations of healthy adult dogs treated with metronidazole. J. Anim. Sci. 2023, 101, skad011. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.H.; Marsilio, S.; Chow, B.; Zornow, K.A.; Slovak, J.E.; Pilla, R.; Lidbury, J.A.; Steiner, J.M.; Park, S.Y.; Hong, M.P.; et al. Dysbiosis index to evaluate the fecal microbiota in healthy cats and cats with chronic enteropathies. J. Feline Med. Surg. 2022, 24, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Verbrugghe, P.; Van Aken, O.; Hallenius, F.; Nilsson, A. Development of a real-time quantitative PCR method for detection and quantification of Prevotella copri. BMC Microbiol. 2021, 21, 23. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Clay, S.L.; Lavoie, S.L.; Bae, S.; Lang, J.K.; Fonseca-Pereira, D.; Rosinski, K.G.; Ou, N.; Glickman, J.N.; Garrett, W.S. Fusobacterium nucleatum drives a pro-inflammatory intestinal microenvironment through metabolite receptor-dependent modulation of IL-17 expression. Gut Microbes 2021, 13, 1987780. [Google Scholar] [CrossRef]
- Pilla, R.; Suchodolski, J.S. The role of the canine gut microbiome and metabolome in health and gastrointestinal disease. Front. Vet. Sci. 2019, 6, 498. [Google Scholar] [CrossRef] [PubMed]
- Weingarden, A.R.; Dosa, P.I.; DeWinter, E.; Steer, C.J.; Shaughnessy, M.K.; Johnson, J.R.; Khoruts, A.; Sadowsky, M.J. Changes in colonic bile acid composition following fecal microbiota transplantation are sufficient to control Clostridium difficile germination and growth. PLoS ONE 2016, 11, e0147210. [Google Scholar] [CrossRef]
- Blake, A.B.; Cigarroa, A.; Klein, H.L.; Khattab, M.R.; Keating, T.; Van De Coevering, P.; Lidbury, J.A.; Steiner, J.M.; Suchodolski, J.S. Developmental stages in microbiota, bile acids, and clostridial species in healthy puppies. J. Vet. Intern. Med. 2020, 34, 2345–2356. [Google Scholar] [CrossRef]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere 2016, 1, e00045-15. [Google Scholar] [CrossRef]
- Thanissery, R.; Winston, J.A.; Theriot, C.M. Inhibition of spore germination, growth, and toxin activity of clinically relevant C. difficile strains by gut microbiota derived secondary bile acids. Anaerobe 2017, 45, 86–100. [Google Scholar] [CrossRef]
- Wang, S.; Martins, R.; Sullivan, M.C.; Friedman, E.S.; Misic, A.M.; El-Fahmawi, A.; De Martinis, E.C.P.; O’Brien, K.; Chen, Y.; Bradley, C.; et al. Diet-induced remission in chronic enteropathy is associated with altered microbial community structure and synthesis of secondary bile acids. Microbiome 2019, 7, 126. [Google Scholar] [CrossRef] [PubMed]
- Guard, B.C.; Honneffer, J.B.; Jergens, A.E.; Jonika, M.M.; Toresson, L.; Lawrence, Y.A.; Webb, C.B.; Hill, S.; Lidbury, J.A.; Steiner, J.M.; et al. Longitudinal assessment of microbial dysbiosis, fecal unconjugated bile acid concentrations, and disease activity in dogs with steroid-responsive chronic inflammatory enteropathy. J. Vet. Intern. Med. 2019, 33, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Blake, A.B.; Guard, B.C.; Honneffer, J.B.; Lidbury, J.A.; Steiner, J.M.; Suchodolski, J.S. Altered microbiota, fecal lactate, and fecal bile acids in dogs with gastrointestinal disease. PLoS ONE 2019, 14, e0224454. [Google Scholar] [CrossRef] [PubMed]
- Manchester, A.C.; Webb, C.B.; Blake, A.B.; Sarwar, F.; Lidbury, J.A.; Steiner, J.M.; Suchodolski, J.S. Long-term impact of tylosin on fecal microbiota and fecal bile acids of healthy dogs. J. Vet. Intern. Med. 2019, 33, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Larouche-Lebel, É.; Loughran, K.A.; Huh, T.P.; Suchodolski, J.S.; Oyama, M.A. Gut dysbiosis and its associations with gut microbiota-derived metabolites in dogs with myxomatous mitral valve disease. Msystems 2021, 6, e00111–e00121. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, Y.; Minamoto, T.; Isaiah, A.; Sattasathuchana, P.; Buono, A.; Rangachari, V.R.; McNeely, I.H.; Lidbury, J.; Steiner, J.M.; Suchodolski, J.S. Fecal short-chain fatty acid concentrations and dysbiosis in dogs with chronic enteropathy. J. Vet. Intern. Med. 2019, 33, 1608–1618. [Google Scholar] [CrossRef]
- Giaretta, P.R.; Suchodolski, J.S.; Jergens, A.E.; Steiner, J.M.; Lidbury, J.A.; Cook, A.K.; Hanifeh, M.; Spillmann, T.; Kilpinen, S.; Syrja, P.; et al. Bacterial Biogeography of the Colon in Dogs With Chronic Inflammatory Enteropathy. Vet. Pathol. 2020, 57, 258–265. [Google Scholar] [CrossRef]
- Pilla, R.; Guard, B.C.; Blake, A.B.; Ackermann, M.; Webb, C.; Hill, S.; Lidbury, J.A.; Steiner, J.M.; Jergens, A.E.; Suchodolski, J.S. Long-term recovery of the fecal microbiome and metabolome of dogs with steroid-responsive enteropathy. Animals 2021, 11, 2498. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef]
- Vandeputte, D.; Kathagen, G.; D’Hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.D. Rarefaction, alpha diversity, and statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Hillmann, B.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Zhu, Q.; Gohl, D.M.; Beckman, K.B.; Knight, R.; Knights, D. Evaluating the information content of shallow shotgun metagenomics. Msystems 2018, 3, e00069-18. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Normal | Minor Changes | Mild to Moderate Changes | Significant Dysbiosis | Total |
|---|---|---|---|---|---|
| Clinically healthy | 66 (85%) | 8 (10%) | 3 (4%) | 1 (1%) | 78 |
| Chronic enteropathy | 52 (36%) | 17 (12%) | 29 (20%) | 48 (33%) | 146 |
| Acute diarrhea | 8 (47%) | 7 (41%) | 1 (6%) | 1 (6%) | 17 |
| Non-gastrointestinal disease | 19 (54%) | 11 (31%) | 5 (14%) | 0 (0%) | 35 |
| On antibiotics | 0 (0%) | 0 (0%) | 3 (15%) | 17 (85%) | 20 |
| Total | 145 | 43 | 41 | 67 | 296 |
| Bacterial Groups | Shannon | p Value | Adjusted p Value | Observed Features | p Value | Adjusted p Value |
|---|---|---|---|---|---|---|
| DI | −0.23 (−0.34 to −0.11) | <0.0001 | 0.0008 | −0.26 (−0.37 to −0.14) | <0.0001 | 0.0008 |
| Faecalibacterium | 0.38 (0.27–0.47) | <0.0001 | 0.0008 | 0.56 (0.47–0.64) | <0.0001 | 0.0008 |
| Fusobacterium | 0.28 (0.16–0.38) | <0.0001 | 0.0008 | 0.36 (0.25–0.46) | <0.0001 | 0.0008 |
| Clostridium hiranonis | 0.16 (0.05–0.28) | 0.005 | 0.04 | 0.26 (0.14–0.37) | <0.0001 | 0.0008 |
| Turicibacter | 0.11 (−0.007 to 0.23) | 0.06 | 0.48 | 0.28 (0.16–0.39) | <0.0001 | 0.0008 |
| Blautia | 0.10 (−0.02 to 0.22) | 0.08 | 0.64 | 0.25 (0.14–0.36) | <0.0001 | 0.0008 |
| Streptococcus | −0.08 (−0.19 to 0.04) | 0.19 | 1.0 | −0.08 (−0.20 to 0.04) | 0.15 | 1.0 |
| Escherichia coli | −0.11 (−0.22 to 0.01) | 0.07 | 0.56 | 0.02 (−0.10–0.13) | 0.79 | 1.0 |
| Compared to Normal Groups | R Value | p Value |
|---|---|---|
| minor changes | 0.19 | 0.001 |
| mild to moderate changes (0 < DI <2) | 0.24 | 0.001 |
| significant dysbiosis (2 < DI < 5) | 0.54 | 0.001 |
| significant dysbiosis (5 < DI < 8) | 0.73 | 0.001 |
| significant dysbiosis (DI > 8) | 0.91 | 0.001 |
| Compared to −5 < DI < −10 | R Value | p Value |
|---|---|---|
| −5 < DI < 0 | −0.01 | 0.56 |
| 0 < DI < 2 | 0.12 | 0.001 |
| 2 < DI < 5 | 0.30 | 0.001 |
| 5 < DI < 8 | 0.65 | 0.001 |
| DI > 8 | 0.89 | 0.001 |
| Bacterial Groups Targeted in DI | Spearman R (n = 296) Rarefaction Depth of 9788 | Spearman R (n = 285) Rarefaction Depth of 100,000 |
|---|---|---|
| Escherichia coli | 0.80 (0.76–0.84) | 0.84 (0.80–0.87) |
| Faecalibacterium | 0.80 (0.75–0.84) | 0.82 (0.78–0.86) |
| Streptococcus | 0.77 (0.71–0.81) | 0.77 (0.72–0.82) |
| Clostridium hiranonis | 0.73 (0.67–0.78) | 0.74 (0.68–0.79) |
| Fusobacterium | 0.71 (0.65–0.76) | 0.78 (0.72–0.82) |
| Turicibacter | 0.65 (0.57–0.71) | 0.72 (0.65–0.77) |
| Blautia | 0.46 (0.36–0.55) | 0.49 (0.39–0.57) |
| Bacterial Taxa | Sequencing Rarefaction 9788 (n = 78) | Sequencing Rarefaction 100,000 (n = 76) | qPCR (n = 78) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Median | Range | UDL 1 (%) | Median | Range | UDL 1 (%) | Median | Range | UDL 1 (%) | |
| Collinsella | 6.1 | 0–83.2 | 1.3 | 5.9 | 0–83.6 | 1.3 | 13.7 | 13.2–15.0 2 | 0 |
| Blautia | 5.9 | 0.4–42.1 | 0 | 6.3 | 0.3–41.9 | 0 | 10.4 | 9.1–11.1 | 0 |
| Bacteroides | 5.4 | 0–63.4 | 33.3 | 4.5 | 0–62.9 | 0 | 6.7 | 3.2–7.9 | 0 |
| R. gnavus | 4 | 0.2–27.0 | 0 | 4.4 | 0.1–26.9 | 0 | 10.5 | 6.0–12.4 2 | 1.3 |
| C. hiranonis | 2.5 | 0.01–36.9 | 0 | 2.5 | 0–36.7 | 0 | 6.3 | 2.5–7.7 | 0 |
| Prevotella copri | 1.6 | 0–73.5 | 20.7 | 1.8 | 0–75.3 | 16.9 | 14 | 9.5–16.8 2 | 9.0 |
| Faecalibacterium | 0.13 | 0–3.7 | 7.7 | 0.13 | 0–4.1 | 1.3 | 6.4 | 3.1–8.0 | 0 |
| Streptococcus | 0.07 | 0–61.8 | 28.2 | 0.07 | 0–61.4 | 6.5 | 7.8 | 1.1–8.7 | 1.3 |
| Lactobacillus | 0.03 | 0–88.5 | 29.5 | 0.02 | 0–88.1 | 10.5 | N/A 3 | N/A | N/A |
| Bifidobacterium | 0.02 | 0–61.9 | 35.9 | 0.01 | 0–62.7 | 13.1 | 4 | 2.1–7.6 | 0 |
| Fusobacterium | 0.02 | 0–44.4 | 34.6 | 0.04 | 0–44.4 | 22.3 | 9.1 | 6.4–10.8 | 0 |
| Turicibacter | 0.01 | 0–2.3 | 33.3 | 0.02 | 0–2.5 | 10.5 | 6.8 | 4.3–9.0 | 0 |
| Escherichia coli | 0 | 0–4.7 | 67.9 | 0 | 0–5.0 | 52.6 | 4.7 | 0.9–7.7 | 3.8 |
| Species | Median (%) | Range (%) |
|---|---|---|
| unclassified species in the family Lachnospiraceae | 5.1 | 0.04–29.7 |
| Ruminococcus gnavus | 4.4 | 0.13–26.9 |
| Clostridium hiranonis | 2.5 | 0.01–36.7 |
| unclassified species in the order Bacteroidales | 0.6 | 0.001–8.1 |
| Clostridium sp. AT4 | 0.6 | 0.001–13.3 |
| unclassified species in the order Clostridiales | 0.5 | 0.03–2.2 |
| unclassified species in the genus Bacteroides | 0.5 | 0.002–17.0 |
| unclassified species in the phylum Firmicutes | 0.3 | 0.02–1.1 |
| unclassified species | 0.3 | 0.01–1.0 |
| Coprococcus sp. HPP0074 | 0.2 | 0.002–1.7 |
| unclassified species in the genus Blautia | 0.2 | 0.02–11.8 |
| Blautia sp. Marseille P3201T | 0.2 | 0.002–1.3 |
| Clostridium glycyrrhizinilyticum | 0.1 | 0.001–0.6 |
| Blautia wexlerae | 0.05 | 0.004–6.096 |
| Blautia massiliensis | 0.04 | 0.001–0.251 |
| Blautia obeum | 0.03 | 0.002–0.848 |
| Fusicatenibacter saccharivorans | 0.01 | 0.001–0.251 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sung, C.-H.; Pilla, R.; Chen, C.-C.; Ishii, P.E.; Toresson, L.; Allenspach-Jorn, K.; Jergens, A.E.; Summers, S.; Swanson, K.S.; Volk, H.; et al. Correlation between Targeted qPCR Assays and Untargeted DNA Shotgun Metagenomic Sequencing for Assessing the Fecal Microbiota in Dogs. Animals 2023, 13, 2597. https://doi.org/10.3390/ani13162597
Sung C-H, Pilla R, Chen C-C, Ishii PE, Toresson L, Allenspach-Jorn K, Jergens AE, Summers S, Swanson KS, Volk H, et al. Correlation between Targeted qPCR Assays and Untargeted DNA Shotgun Metagenomic Sequencing for Assessing the Fecal Microbiota in Dogs. Animals. 2023; 13(16):2597. https://doi.org/10.3390/ani13162597
Chicago/Turabian StyleSung, Chi-Hsuan, Rachel Pilla, Chih-Chun Chen, Patricia Eri Ishii, Linda Toresson, Karin Allenspach-Jorn, Albert E. Jergens, Stacie Summers, Kelly S. Swanson, Holger Volk, and et al. 2023. "Correlation between Targeted qPCR Assays and Untargeted DNA Shotgun Metagenomic Sequencing for Assessing the Fecal Microbiota in Dogs" Animals 13, no. 16: 2597. https://doi.org/10.3390/ani13162597
APA StyleSung, C.-H., Pilla, R., Chen, C.-C., Ishii, P. E., Toresson, L., Allenspach-Jorn, K., Jergens, A. E., Summers, S., Swanson, K. S., Volk, H., Schmidt, T., Stuebing, H., Rieder, J., Busch, K., Werner, M., Lisjak, A., Gaschen, F. P., Belchik, S. E., Tolbert, M. K., ... Suchodolski, J. S. (2023). Correlation between Targeted qPCR Assays and Untargeted DNA Shotgun Metagenomic Sequencing for Assessing the Fecal Microbiota in Dogs. Animals, 13(16), 2597. https://doi.org/10.3390/ani13162597