
Copy Number Variation Discovery in South African Nguni-Sired and Bonsmara-Sired Crossbred Cattle
 ,
,
Simple Summary
Abstract
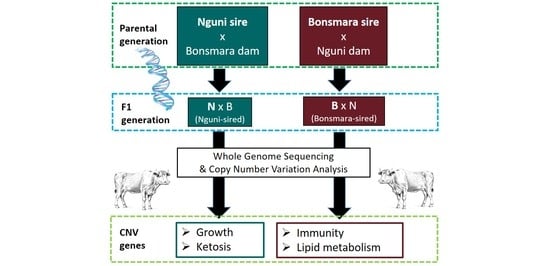
1. Introduction
2. Materials and Methods
2.1. Animal Selection
2.2. DNA Extraction and Sequencing
2.3. Copy Number Variation Analysis
3. Results
3.1. Copy Number Variants Summary Statistics
3.2. CNV Gene Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Habtamu, A.G.; Min, C.; Ping, Y. Applications of Genomic Copy Number Variations on Livestock: A Review. Afr. J. Biotechnol. 2018, 17, 1313–1323. [Google Scholar] [CrossRef]
- Keel, B.N.; Lindholm-Perry, A.K.; Snelling, W.M. Evolutionary and Functional Features of Copy Number Variation in the Cattle Genome. Front. Genet. 2016, 7, 207. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hou, Y.; Bickhart, D.M.; Zhou, Y.; Hay, E.H.A.; Song, J.; Sonstegard, T.S.; Van Tassell, C.P.; Liu, G.E. Population-Genetic Properties of Differentiated Copy Number Variations in Cattle. Sci. Rep. 2016, 6, 23161. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.E.; Hou, Y.; Zhu, B.; Cardone, M.F.; Jiang, L.; Cellamare, A.; Mitra, A.; Alexander, L.J.; Coutinho, L.L.; Dell’Aquila, M.E.; et al. Analysis of Copy Number Variations among Diverse Cattle Breeds. Genome Res. 2010, 20, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Bhanuprakash, V.; Chhotaray, S.; Pruthviraj, D.R.; Rawat, C.; Karthikeyan, A.; Panigrahi, M. Copy Number Variation in Livestock: A Mini Review. Vet. World 2018, 11, 535–541. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, Y.; Wang, X.; Zhang, Y.; Zhao, X. Identification of Copy Number Variations in Qinchuan Cattle Using BovineHD Genotyping Beadchip Array. Mol. Genet. Genom. 2015, 290, 319–327. [Google Scholar] [CrossRef]
- Wang, M.D.; Dzama, K.; Hefer, C.A.; Muchadeyi, F.C. Genomic Population Structure and Prevalence of Copy Number Variations in South African Nguni Cattle. BMC Genom. 2015, 16, 894. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, S.; Yang, M.; Xu, Y.; Li, C.; Sun, J.; Huang, Y.; Lan, X.; Lei, C.; Zhou, Y.; et al. Detection of Copy Number Variations and Their Effects in Chinese Bulls. BMC Genom. 2014, 15, 480. [Google Scholar] [CrossRef]
- Liu, M.; Fang, L.; Liu, S.; Pan, M.G.; Seroussi, E.; Cole, J.B.; Ma, L.; Chen, H.; Liu, G.E. Array CGH-Based Detection of CNV Regions and Their Potential Association with Reproduction and Other Economic Traits in Holsteins. BMC Genom. 2019, 20, 181. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Cardone, M.F.; Wang, K.; Kim, E.-S.; Matukumalli, L.K.; Ventura, M.; Song, J.; VanRaden, P.M.; et al. Genomic Characteristics of Cattle Copy Number Variations. BMC Genom. 2011, 12, 127. [Google Scholar] [CrossRef]
- Prayaga, K.C.; Henshall, J.M. Adaptability in Tropical Beef Cattle: Genetic Parameters of Growth, Adaptive and Temperament Traits in a Crossbred Population. Aust. J. Exp. Agric. 2005, 45, 971–983. [Google Scholar] [CrossRef]
- Mote, S.S.; Chauhan, D.S.; Ghosh, N. Effect of Environment Factors on Milk Production and Lactation Length under Different Seasons in Crossbred Cattle. Indian J. Anim. Res. 2016, 50, 175–180. [Google Scholar] [CrossRef][Green Version]
- Antunes de Lemos, M.V.; Berton, M.P.; Ferreira de Camargo, G.M.; Peripolli, E.; de Oliveira Silva, R.M.; Ferreira Olivieri, B.; Cesar, A.S.M.; Pereira, A.S.C.; de Albuquerque, L.G.; de Oliveira, H.N.; et al. Copy Number Variation Regions in Nellore Cattle: Evidences of Environment Adaptation. Livest. Sci. 2018, 207, 51–58. [Google Scholar] [CrossRef]
- Pyoos, G. Crossbreeding Effects of Cow Efficiency and Component Traits. Ph.D. Thesis, University of the Free State, Bloemfontein, South Africa, 2018. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Hou, Y.; Schroeder, S.G.; Alkan, C.; Cardone, M.F.; Matukumalli, L.K.; Song, J.; Schnabel, R.D.; Ventura, M.; Taylor, J.F.; et al. Copy Number Variation of Individual Cattle Genomes Using Next-Generation Sequencing. Genome Res. 2012, 22, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Mielczarek, M.; Fraszczak, M.; Nicolazzi, E.; Williams, J.L.; Szyda, J. Landscape of Copy Number Variations in Bos taurus: Individual- and Inter-Breed Variability. BMC Genom. 2018, 19, 410. [Google Scholar] [CrossRef]
- Zhan, B.; Fadista, J.; Thomsen, B.; Hedegaard, J.; Panitz, F.; Bendixen, C. Global Assessment of Genomic Variation in Cattle by Genome Resequencing and High-Throughput Genotyping. BMC Genom. 2011, 12, 557. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Choi, J.-W.; Basu, U.; Sumner-Thomson, J.M.; Meng, Y.; Liao, X.; Moore, S.S. Whole Genome Resequencing of Black Angus and Holstein Cattle for SNP and CNV Discovery. BMC Genom. 2011, 12, 559. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-W.; Liao, X.; Stothard, P.; Chung, W.-H.; Jeon, H.-J.; Miller, S.P.; Choi, S.-Y.; Lee, J.-K.; Yang, B.; Lee, K.-T. Whole-Genome Analyses of Korean Native and Holstein Cattle Breeds by Massively Parallel Sequencing. PLoS ONE 2014, 9, e101127. [Google Scholar] [CrossRef]
- Guo, S.; Wu, X.; Pei, J.; Wang, X.; Bao, P.; Xiong, L.; Chu, M.; Liang, C.; Yan, P.; Guo, X. Genome-Wide CNV Analysis Reveals Variants Associated with High-Altitude Adaptation and Meat Traits in Qaidam Cattle. Electron. J. Biotechnol. 2021, 54, 8–16. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Canavez, F.C.; Luche, D.D.; Stothard, P.; Leite, K.R.M.; Sousa-Canavez, J.M.; Plastow, G.; Meidanis, J.; Souza, M.A.; Feijao, P.; Moore, S.S. Genome Sequence and Assembly of Bos Indicus. J. Hered. 2012, 103, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Van Marle-Köster, E.; Visser, C.; Sealy, J.; Frantz, L. Capitalizing on the Potential of South African Indigenous Beef Cattle Breeds: A Review. Sustainability 2021, 13, 4388. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup 1000 Genome Project Data Processing the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Povysil, G.; Tzika, A.; Vogt, J.; Haunschmid, V.; Messiaen, L.; Zschocke, J.; Klambauer, G.; Hochreiter, S.; Wimmer, K. Panelcn.MOPS: Copy-Number Detection in Targeted NGS Panel Data for Clinical Diagnostics. Hum. Mutat. 2017, 38, 889–897. [Google Scholar] [CrossRef]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clevert, D.A.; Mitterecker, A.; Bodenhofer, U.; Hochreiter, S.C. MOPS: Mixture of Poissons for Discovering Copy Number Variations in next-Generation Sequencing Data with a Low False Discovery Rate. Nucleic Acids Res. 2012, 40, e69. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Shin, J.M.; Yoo, K.J.; Kim, M.S.; Kim, D.; Baek, K.H. Hyaluronan- and RNA-Binding Deubiquitinating Enzymes of USP17 Family Members Associated with Cell Viability. BMC Genom. 2006, 7, 292. [Google Scholar] [CrossRef]
- Sinha, M.K.; Kumaresan, A.; Rao Talluri, T.; Peter, J.; King, E.S.; Prakash, M.A.; Nag, P.; Paul, N.; Raval, K.; Kamaraj, E. SNPs Cumulating to Genetic Variation for Fertility in Crossbred (Bos taurus × Bos indicus) Bull Spermatozoa. Res. Sq. 2022. preprint. [Google Scholar] [CrossRef]
- Cruz, V.A.R.; Oliveira, H.R.; Brito, L.F.; Fleming, A.; Larmer, S.; Miglior, F.; Schenkel, F.S. Genome-Wide Association Study for Milk Fatty Acids in Holstein Cattle Accounting for the Dgat1 Gene Effect. Animals 2019, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Nguyen, D.T.; Choi, M.; Cha, S.Y.; Kim, J.H.; Dadi, H.; Seo, H.G.; Seo, K.; Chun, T.; Park, C. Analysis of Cattle Olfactory Subgenome: The First Detail Study on the Characteristics of the Complete Olfactory Receptor Repertoire of a Ruminant. BMC Genom. 2013, 14, 596. [Google Scholar] [CrossRef] [PubMed]
- Harima, Y.; Takashima, Y.; Ueda, Y.; Ohtsuka, T.; Kageyama, R. Accelerating the Tempo of the Segmentation Clock by Reducing the Number of Introns in the Hes7 Gene. Cell Rep. 2013, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kominakis, A.; Hager-Theodorides, A.L.; Zoidis, E.; Saridaki, A.; Antonakos, G.; Tsiamis, G. Combined GWAS and ‘Guilt by Association’-Based Prioritization Analysis Identifies Functional Candidate Genes for Body Size in Sheep. Genet. Sel. Evol. 2017, 49, 41. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, H.; Rafat, S.A.; Shodja, J.; Moradi, M.H.; Moradi Shahrebabak, H. Genome-Wide Association Study and Gene Ontology for Growth and Wool Characteristics in Zandi Sheep. J. Livest. Sci. Technol. 2020, 8, 45–55. [Google Scholar] [CrossRef]
- Behl, J.D.; Verma, N.K.; Tyagi, N.; Mishra, P.; Behl, R.; Joshi, B.K. The Major Histocompatibility Complex in Bovines: A Review. Int. Sch. Res. Not. 2012, 2012, 872710. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, M.; da Silva, V.H.; Megens, H.J.; Visker, M.H.P.W.; Ajmone-Marsan, P.; Bâlteanu, V.A.; Dunner, S.; Garcia, J.F.; Ginja, C.; Kantanen, J.; et al. Distribution and Functionality of Copy Number Variation across European Cattle Populations. Front. Genet. 2017, 8, 108. [Google Scholar] [CrossRef]
- Agerholm, J.S.; McEvoy, F.J.; Heegaard, S.; Charlier, C.; Jagannathan, V.; Drögemüller, C. A de Novo Missense Mutation of FGFR2 Causes Facial Dysplasia Syndrome in Holstein Cattle. BMC Genet. 2017, 18, 74. [Google Scholar] [CrossRef]
- Maciel Jr, V.L.; Caldas-Bussiere, M.C.; Silveira, V.; Reis, R.S.; Rios, A.F.L.; de Carvalho, C.S.P. L-Arginine Alters the Proteome of Frozen-Thawed Bovine Sperm during in Vitro Capacitation. Theriogenology 2018, 119, 1–9. [Google Scholar] [CrossRef]
- Klein, S.L.; Scheper, C.; Brügemann, K.; Swalve, H.H.; König, S. Phenotypic Relationships, Genetic Parameters, Genome-Wide Associations, and Identification of Potential Candidate Genes for Ketosis and Fat-to-Protein Ratio in German Holstein Cows. J. Dairy Sci. 2019, 102, 6276–6287. [Google Scholar] [CrossRef]
- Mei, C.; Junjvlieke, Z.; Raza, S.H.A.; Wang, H.; Cheng, G.; Zhao, C.; Zhu, W.; Zan, L. Copy Number Variation Detection in Chinese Indigenous Cattle by Whole Genome Sequencing. Genomics 2020, 112, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fan, H.; Jing, S.; Xia, J.; Chen, Y.; Zhang, L.; Gao, X.; Li, J.; Gao, H.; Ren, H. A Genome-wide Scan for Copy Number Variations Using High-density Single Nucleotide Polymorphism Array in Simmental Cattle. Anim. Genet. 2015, 46, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Keel, B.N.; Keele, J.W.; Snelling, W.M. Genome-Wide Copy Number Variation in the Bovine Genome Detected Using Low Coverage Sequence of Popular Beef Breeds. Anim. Genet. 2017, 48, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of Change in Gene Copy Number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.D.; Dzama, K.; Muchadeyi, F.C. Genetic Diversity of Seven Cattle Breeds Inferred Using Copy Number Variations. Front. Genet. 2018, 9, 163. [Google Scholar] [CrossRef]
- Turner, D.J.; Miretti, M.; Rajan, D.; Fiegler, H.; Carter, N.P.; Blayney, M.L.; Beck, S.; Hurles, M.E. Germline Rates of de Novo Meiotic Deletions and Duplications Causing Several Genomic Disorders. Nat. Genet. 2008, 40, 90–95. [Google Scholar] [CrossRef]
- Higgins, M.G.; Kenny, D.A.; Fitzsimons, C.; Blackshields, G.; Coyle, S.; McKenna, C.; McGee, M.; Morris, D.W.; Waters, S.M. The Effect of Breed and Diet Type on the Global Transcriptome of Hepatic Tissue in Beef Cattle Divergent for Feed Efficiency. BMC Genom. 2019, 20, 525. [Google Scholar] [CrossRef]
- Terry, S.A.; Basarab, J.A.; Guan, L.L.; McAllister, T.A. Strategies to Improve the Efficiency of Beef Cattle Production. Can. J. Anim. Sci. 2020, 101, 1–19. [Google Scholar] [CrossRef]
- Lui, J.C.; Baron, J. Mechanisms Limiting Body Growth in Mammals. Endocr. Rev. 2011, 32, 422–440. [Google Scholar] [CrossRef]
- Liu, G.E.; Brown, T.; Hebert, D.A.; Cardone, M.F.; Hou, Y.; Choudhary, R.K.; Shaffer, J.; Amazu, C.; Connor, E.E.; Ventura, M.; et al. Initial Analysis of Copy Number Variations in Cattle Selected for Resistance or Susceptibility to Intestinal Nematodes. Mamm. Genome 2011, 22, 111–121. [Google Scholar] [CrossRef]
- le Roex, N.; Koets, A.P.; van Helden, P.D.; Hoal, E.G. Gene Polymorphisms in African Buffalo Associated with Susceptibility to Bovine Tuberculosis Infection. PLoS ONE 2013, 8, e64494. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xu, J.; Liu, X.; Zhang, Z.; Zhong, J.; Wen, Y.; Yao, Z.; Yang, P.; Wang, E.; Chen, F. The Copy Number Variation of DMBT1 Gene Effects Body Traits in Two Chinese Cattle Breeds. 3 Biotech 2022, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Touhara, K.; Vosshall, L.B. Sensing Odorants and Pheromones with Chemosensory Receptors. Annu. Rev. Physiol. 2009, 71, 307–332. [Google Scholar] [CrossRef] [PubMed]
- Kerley, G.I.H.; Behrens, K.G.; Carruthers, J.; Diemont, M.; Du Plessis, J.; Minnie, L.; Richardson, P.R.K.; Somers, M.J.; Tambling, C.J.; Turpie, J. Livestock Predation in South Africa: The Need for and Value of a Scientific Assessment. S. Afr. J. Sci. 2017, 113, 3. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| BTA No. | Chr Length (Mb) | Cumulative CNV Length | No. of CNVs | Mean Length (bp) | Median Length (bp) | Min Length (bp) | Max Length (bp) |
|---|---|---|---|---|---|---|---|
| 1 | 161.11 | 1899 | 3 | 633 | 314 | 314 | 1271 |
| 2 | 140.68 | 12,375 | 7 | 1768 | 1201 | 746 | 4987 |
| 3 | 127.87 | 86,703 | 10 | 8670.3 | 2597 | 515 | 36,426 |
| 4 | 124.43 | 76,137 | 12 | 6345 | 3254 | 749 | 21,076 |
| 5 | 125.64 | 334,329 | 25 | 13,373 | 2858 | 935 | 96,975 |
| 6 | 122.65 | 52,759 | 11 | 4796 | 3236 | 1007 | 13,724 |
| 7 | 111.95 | 61,680 | 15 | 4112 | 2435 | 410 | 16,115 |
| 8 | 116.94 | 753,177 | 24 | 31,382 | 4920 | 230 | 110,871 |
| 9 | 108.1 | 25,279 | 7 | 3611 | 2357 | 896 | 9901 |
| 10 | 106.31 | 68,741 | 11 | 6249 | 959 | 428 | 56,532 |
| 11 | 110.26 | 263,313 | 17 | 15,489 | 2908 | 785 | 70,362 |
| 12 | 85.44 | 50,569 | 3 | 16,856 | 16,673 | 2637 | 31,259 |
| 13 | 84.43 | 32,268 | 10 | 3227 | 2570 | 1418 | 6174 |
| 14 | 81.41 | 8512 | 7 | 1216 | 1269 | 751 | 1464 |
| 15 | 84.8 | 380,029 | 37 | 10,271 | 962 | 929 | 274,172 |
| 16 | 77.91 | 12,954 | 5 | 2591 | 2583 | 1341 | 3674 |
| 17 | 76.52 | 25,833 | 9 | 2870 | 1682 | 1005 | 6364 |
| 18 | 65.95 | 231,509 | 26 | 8904 | 3256 | 575 | 101,156 |
| 19 | 65.32 | 246,360 | 27 | 9124 | 2473 | 296 | 164,086 |
| 20 | 75.86 | 86,586 | 3 | 28,862 | 18,014 | 7797 | 60,775 |
| 21 | 69.31 | 10,296 | 7 | 1471 | 1286 | 899 | 2081 |
| 22 | 61.89 | 48,558 | 12 | 4047 | 2478 | 837 | 11,298 |
| 23 | 53.33 | 188,807 | 15 | 12,587 | 1925 | 311 | 54,452 |
| 24 | 65.02 | 14,363 | 2 | 7182 | 7181.5 | 6451 | 7912 |
| 25 | 44.04 | 405,962 | 22 | 18,453 | 4102 | 968 | 153,354 |
| 26 | 51.86 | 118,236 | 3 | 39,412 | 58,321 | 1594 | 58,321 |
| 27 | 48.75 | 13,346 | 1 | - | - | - | - |
| 28 | 46.11 | 3465 | 3 | 1155 | 1231 | 374 | 1860 |
| 29 | 52.13 | 40,348 | 13 | 3104 | 1214 | 887 | 10,024 |
| X | 88.52 | 19,726 | 8 | 2466 | 1710.5 | 605 | 8804 |
| Total | 2634.54 | 3,674,119 | 355 | 9318 | 2478 | 230 | 274,172 |
| Sample | Function | BTA | CN | CNV Start | CNV End | Gene Start | Gene End | Gene | Full Name | References |
|---|---|---|---|---|---|---|---|---|---|---|
| BXN 3 + 4 | GDP Binding | X | CN1 | 33981816 | 33982421 | 33975376 | 33985271 | RAB9B | RAB9B, member RAS oncogene family | |
| BXN 4 + 5 | Regulation of cellular processes | 3 | CN1 | 126931388 | 126932989 | 126930672 | 126933578 | LOC109556616 | ubiquitin carboxyl-terminal hydrolase 17-like protein 6 | [30] |
| BXN 4 + 5 | Fertility-related factors | 7 | CN1 | 15428095 | 15428909 | 15428030 | 15429198 | RPS28 | ribosomal protein S28 | [31] |
| BXN 4 + 5 | 9 | CN1 | 106343904 | 106344800 | 106342953 | 106345181 | LOC109563685 | probable plastid-lipid-associated protein 3, chloroplastic | ||
| BXN 4 + 5 | Lipid metabolism | 14 | CN1 | 564233 | 565697 | 563768 | 566833 | MAF1 | MAF1 homolog, negative regulator of RNA polymerase III | [32] |
| BXN 4 + 5 | Olfaction | 15 | CN1 | 80006277 | 80007209 | 80006277 | 80007209 | LOC109569114 | olfactory receptor 8J1-like | [33] |
| BXN 4 + 5 | Protein binding | 17 | CN1 | 55217115 | 55218797 | 55216878 | 55219698 | ARL6IP4 | ADP ribosylation factor like GTPase 6 interacting protein 4 | |
| BXN 4 + 5 | Body size | 19 | CN1 | 28307250 | 28309723 | 28306270 | 28309725 | HES7 | hes family bHLH transcription factor 7 | [34,35] |
| BXN 4 + 5 | Growth | 19 | CN1 | 39865977 | 39869076 | 39865484 | 39869235 | SCRN2 | secernin 2 | [36] |
| BXN 4 + 5 | Immunity | 19 | CN1 | 58635110 | 58637647 | 58635110 | 58637827 | LOC109573358 | CMRF35-like molecule 6 | |
| BXN 4 + 5 | Adaptive immunity | 23 | CN1 | 26144727 | 26199179 | 26144339 | 26199240 | LOC109577039 | SLA class II histocompatibility antigen, DQ haplotype D alpha chain | [37] |
| BXN 4 + 5 | Inflammatory response | 25 | CN1 | 10600555 | 10645081 | 10600419 | 10646357 | CIITA | class II major histocompatibility complex transactivator | [38] |
| BXN 4 + 5 | Disease | 26 | CN1 | 43114715 | 43173036 | 43114701 | 43173429 | DMBT1 | deleted in malignant brain tumors 1 | [39] |
| Sample | Function | BTA | CN | CNV Start | CNV End | Gene Start | Gene End | Gene | Full Name | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| NxB 1 + 2 | Fertility-related factors | 8 | CN3 | 7212767 | 7215055 | 7207894 | 7215055 | LOC109562432 | disintegrin and metalloproteinase domain-containing protein 20-like | [40] |
| NxB 1 + 2 | Ketosis | 8 | CN0 | 9713354 | 9824225 | 9711258 | 9901658 | HMBOX1 | homeobox containing 1 | [41] |
| NxB 1 + 2 | Amino acid transport, Growth | 18 | CN1 | 61411652 | 61418513 | 61411629 | 61418610 | LOC109572916 | cationic amino acid transporter 3-like | [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kooverjee, B.B.; Soma, P.; van der Nest, M.A.; Scholtz, M.M.; Neser, F.W.C. Copy Number Variation Discovery in South African Nguni-Sired and Bonsmara-Sired Crossbred Cattle. Animals 2023, 13, 2513. https://doi.org/10.3390/ani13152513
Kooverjee BB, Soma P, van der Nest MA, Scholtz MM, Neser FWC. Copy Number Variation Discovery in South African Nguni-Sired and Bonsmara-Sired Crossbred Cattle. Animals. 2023; 13(15):2513. https://doi.org/10.3390/ani13152513
Chicago/Turabian StyleKooverjee, Bhaveni B., Pranisha Soma, Magrieta A. van der Nest, Michiel M. Scholtz, and Frederick W. C. Neser. 2023. "Copy Number Variation Discovery in South African Nguni-Sired and Bonsmara-Sired Crossbred Cattle" Animals 13, no. 15: 2513. https://doi.org/10.3390/ani13152513
APA StyleKooverjee, B. B., Soma, P., van der Nest, M. A., Scholtz, M. M., & Neser, F. W. C. (2023). Copy Number Variation Discovery in South African Nguni-Sired and Bonsmara-Sired Crossbred Cattle. Animals, 13(15), 2513. https://doi.org/10.3390/ani13152513