
RNA-Seq Analysis of Peripheral Whole Blood from Dairy Bulls with High and Low Antibody-Mediated Immune Responses—A Preliminary Study
 , , , , , , and
, , , , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
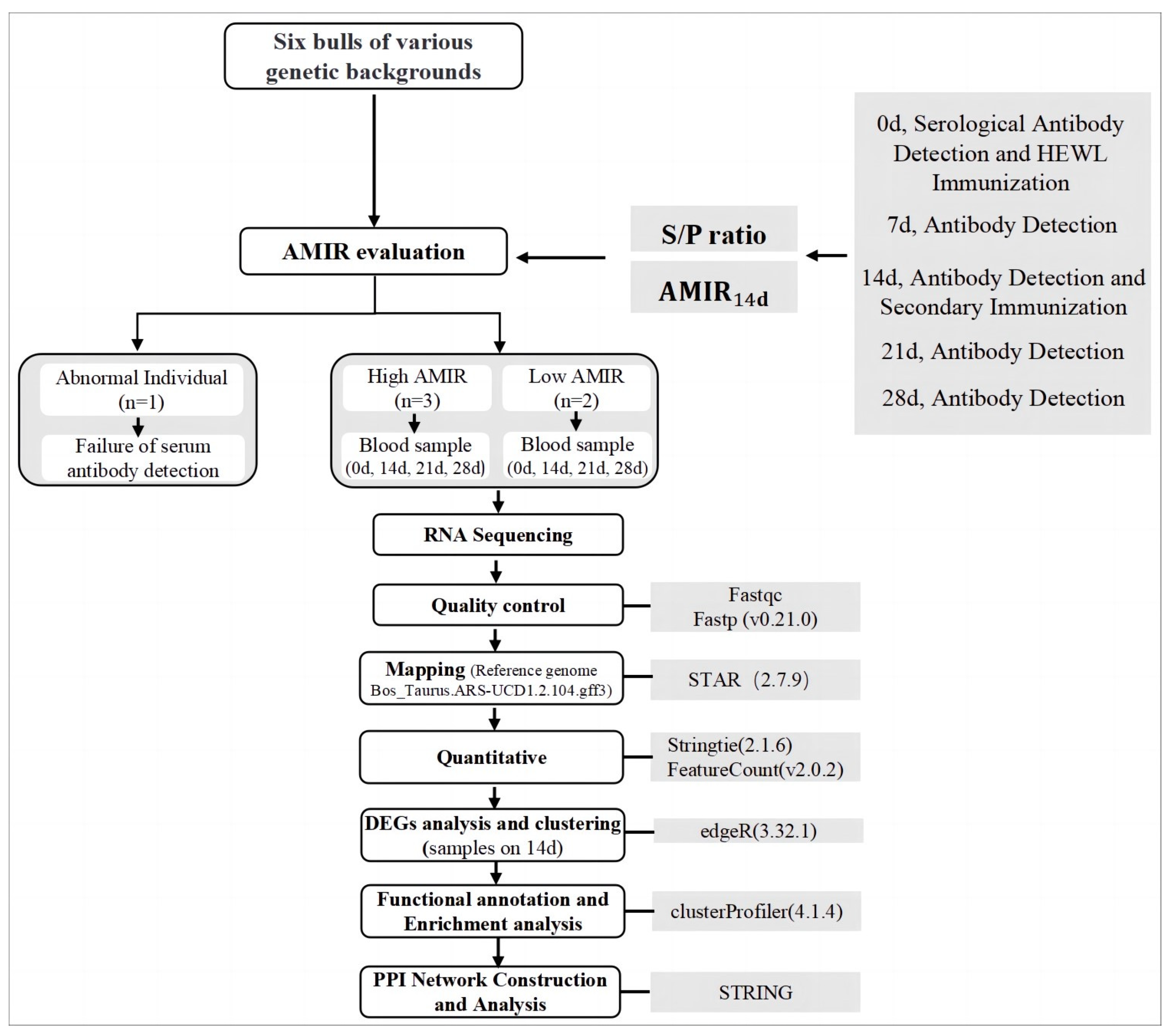
2. Materials and Methods
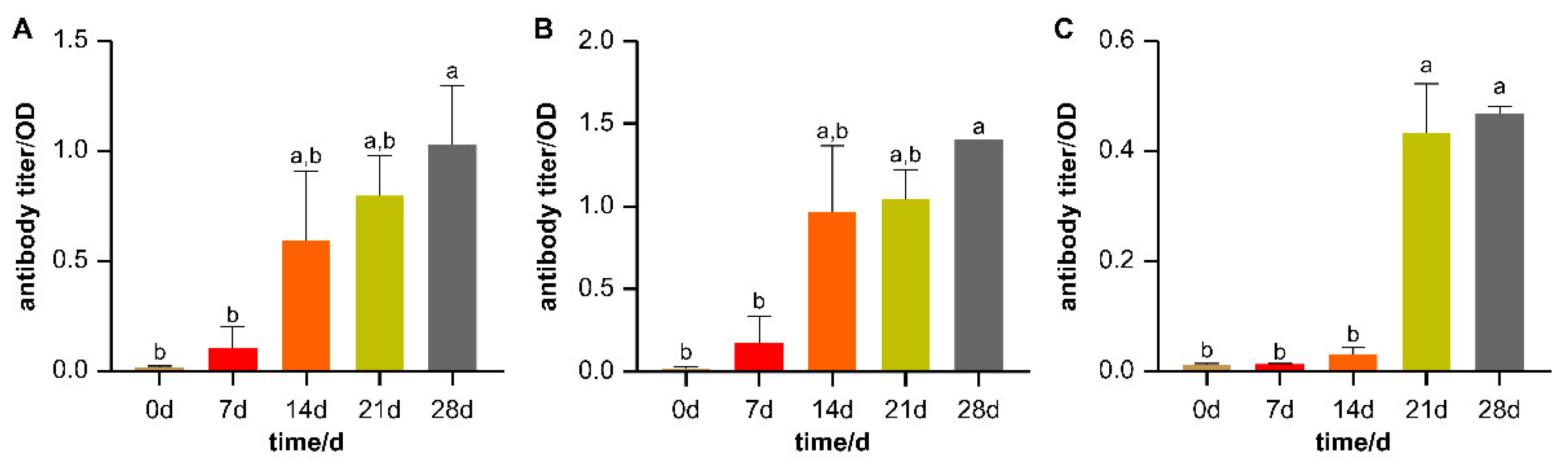
2.1. Experimental Animals and Immunization Protocol
2.2. HEWL Serum Antibody Detection
2.3. RNA-Seq and Transcriptome Quantification
2.4. Analysis of DEGs
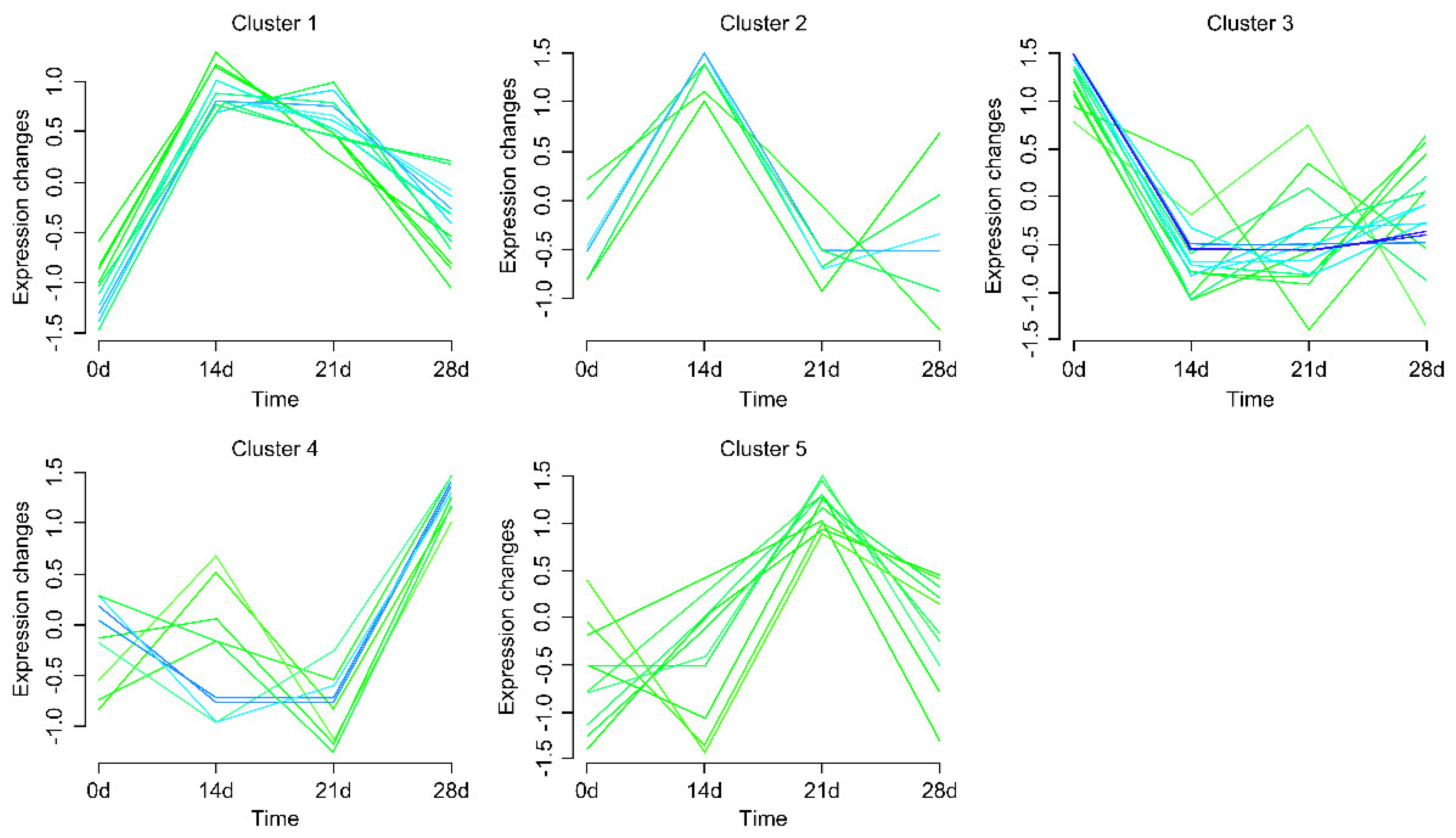
2.5. Gene Expression Pattern Profiling
3. Results
3.1. Experimental Animals and Grouping
3.2. Differentially Expressed Genes
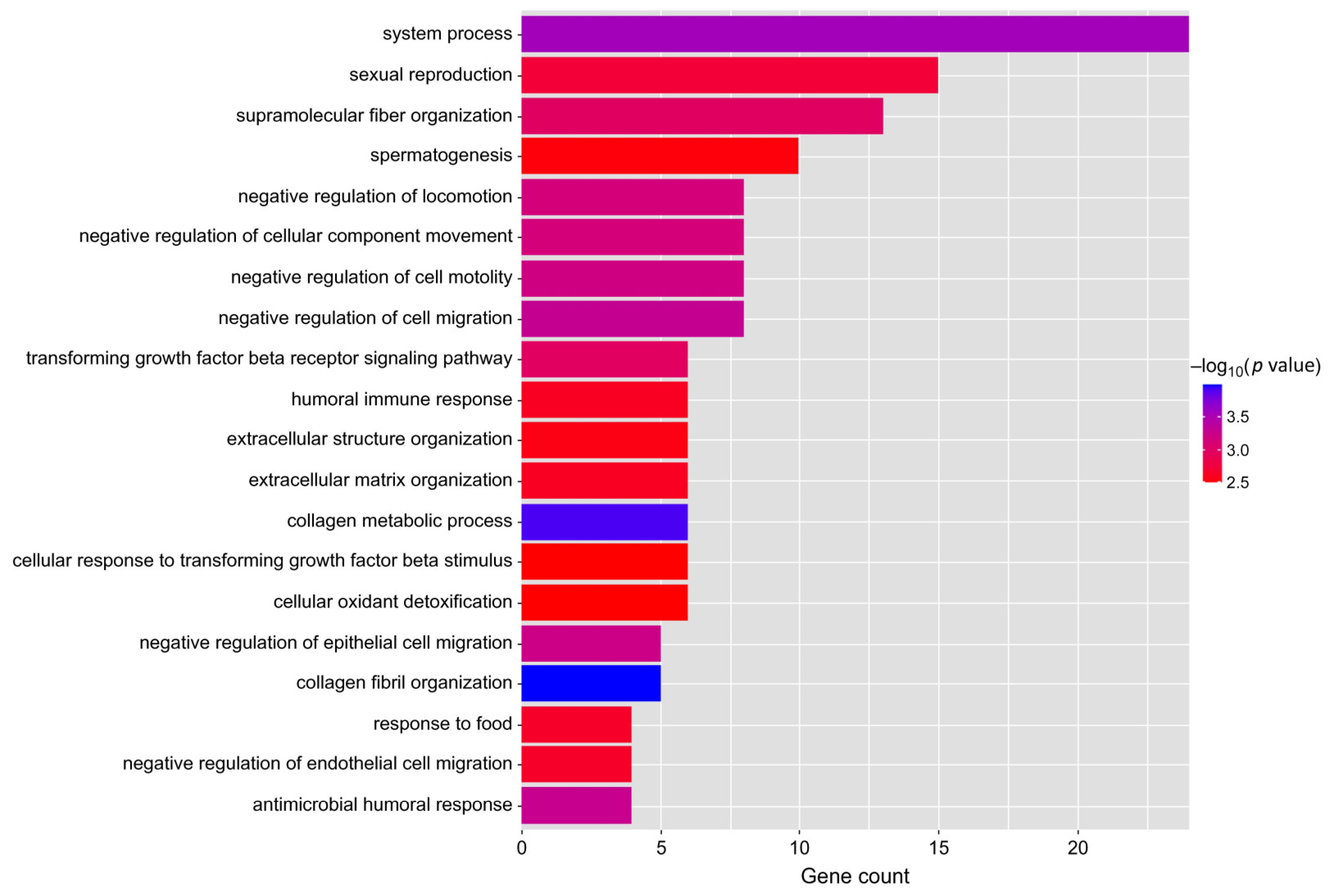
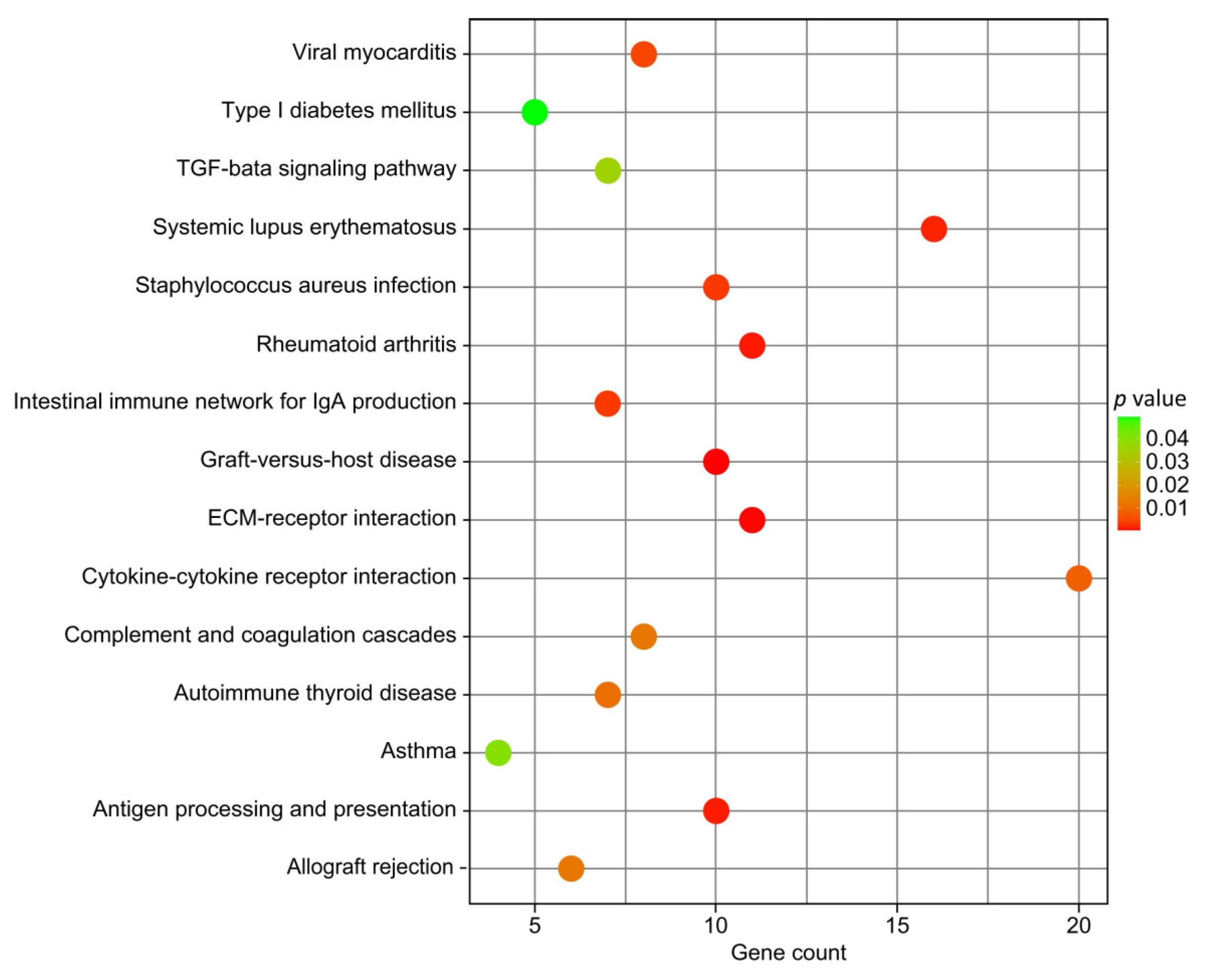
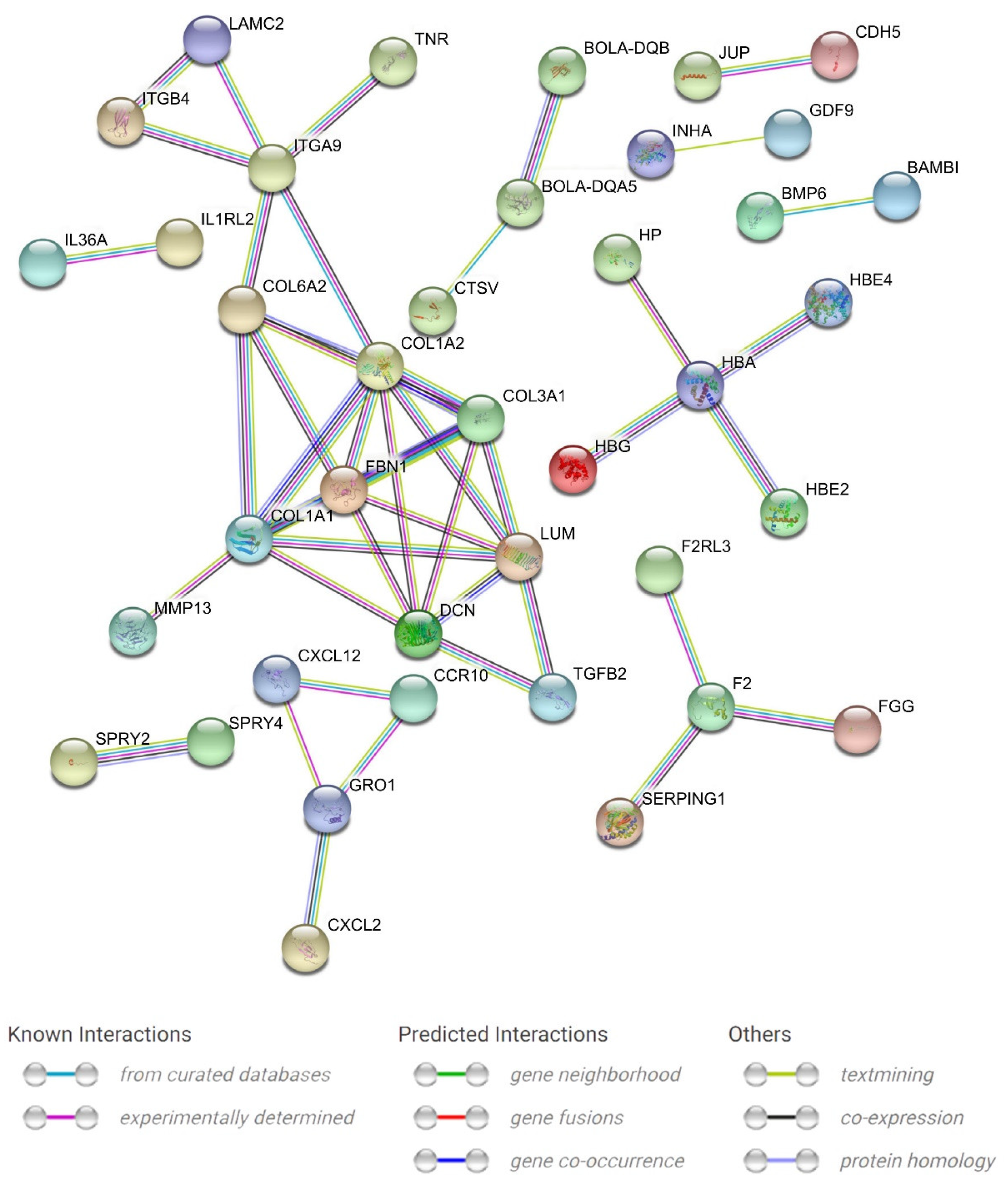
3.3. Functional Enrichment Analysis
3.4. Expression Pattern of Selected DEGs
4. Discussion
4.1. Experimental Design and Samples Information
4.2. Differential Genes Screening and Functional Enrichment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barkema, H.W.; von Keyserlingk, M.A.G.; Kastelic, J.P.; Lam, T.J.G.M.; Luby, C.; Roy, J.-P.; LeBlanc, S.J.; Keefe, G.P.; Kelton, D.F. Invited Review: Changes in the Dairy Industry Affecting Dairy Cattle Health and Welfare. J. Dairy Sci. 2015, 98, 7426–7445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeck, A.; Miglior, F.; Jamrozik, J.; Kelton, D.F.; Schenkel, F.S. Genetic Associations of Ketosis and Displaced Abomasum with Milk Production Traits in Early First Lactation of Canadian Holsteins. J. Dairy Sci. 2013, 96, 4688–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oltenacu, P.; Broom, D. The Impact of Genetic Selection for Increased Milk Yield on the Welfare of Dairy Cows. Anim. Welf. 2010, 19, 39–49. [Google Scholar] [CrossRef]
- Gaddis, K.P. Development of National Genomic Evaluations for Health Traits in U.S. Holsteins. In Proceedings of the World Congress of Genetics Applied in Livestock Production, Auckland, New Zealand, 11–16 February 2018; p. 594. [Google Scholar]
- Egger-Danner, C.; Cole, J.B.; Pryce, J.E.; Gengler, N.; Heringstad, B.; Bradley, A.; Stock, K.F. Invited Review: Overview of New Traits and Phenotyping Strategies in Dairy Cattle with a Focus on Functional Traits. Animal 2015, 9, 191–207. [Google Scholar] [CrossRef] [Green Version]
- Thompson-Crispi, K.A.; Sewalem, A.; Miglior, F.; Mallard, B.A. Genetic Parameters of Adaptive Immune Response Traits in Canadian Holsteins. J. Dairy Sci. 2012, 95, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Heriazon, A.; Quinton, M.; Miglior, F.; Leslie, K.E.; Sears, W.; Mallard, B.A. Phenotypic and Genetic Parameters of Antibody and Delayed-Type Hypersensitivity Responses of Lactating Holstein Cows. Vet. Immunol. Immunopathol. 2013, 154, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Thompson-Crispi, K.; Atalla, H.; Miglior, F.; Mallard, B.A. Bovine Mastitis: Frontiers in Immunogenetics. Front. Immunol. 2014, 5, 493. [Google Scholar] [CrossRef] [PubMed]
- Thompson-Crispi, K.A.; Miglior, F.; Mallard, B.A. Incidence Rates of Clinical Mastitis among Canadian Holsteins Classified as High, Average, or Low Immune Responders. Clin. Vaccine Immunol. 2013, 20, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Mallard, B.A.; Emam, M.; Paibomesai, M.; Thompson-Crispi, K.; Wagter-Lesperance, L. Genetic Selection of Cattle for Improved Immunity and Health. Jpn. J. Vet. Res. 2015, 63, S37–S44. [Google Scholar]
- Fang, L.; Sahana, G.; Ma, P.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.S.; Sørensen, P. Exploring the Genetic Architecture and Improving Genomic Prediction Accuracy for Mastitis and Milk Production Traits in Dairy Cattle by Mapping Variants to Hepatic Transcriptomic Regions Responsive to Intra-Mammary Infection. Genet. Sel. Evol. 2017, 49, 44. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Sahana, G.; Su, G.; Yu, Y.; Zhang, S.; Lund, M.; Sørensen, P. Integrating Sequence-Based GWAS and RNA-Seq Provides Novel Insights into the Genetic Basis of Mastitis and Milk Production in Dairy Cattle. Sci. Rep. 2017, 7, 45560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Huang, H.; Freebern, E.; Santos, D.J.A.; Dai, D.; Si, J.; Ma, C.; Cao, J.; Guo, G.; Liu, G.E.; et al. Integrating RNA-Seq with GWAS Reveals Novel Insights into the Molecular Mechanism Underpinning Ketosis in Cattle. BMC Genom. 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Prasad, M.; Mishra, B.; Manjunath, S.; Sahu, A.R.; Bhuvana Priya, G.; Wani, S.A.; Sahoo, A.P.; Kumar, A.; Balodi, S.; et al. Transcriptome Analysis Reveals Common Differential and Global Gene Expression Profiles in Bluetongue Virus Serotype 16 (BTV-16) Infected Peripheral Blood Mononuclear Cells (PBMCs) in Sheep and Goats. Genom. Data 2017, 11, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Barreto, D.M.; Barros, G.S.; Santos, L.A.B.O.; Soares, R.C.; Batista, M.V.A. Comparative Transcriptomic Analysis of Bovine Papillomatosis. BMC Genom. 2018, 19, 949. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Y.; Liang, L.; Cui, S.; Zhang, Y. RNA-Seq Based Transcriptome Analysis during Bovine Viral Diarrhoea Virus (BVDV) Infection. BMC Genom. 2019, 20, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, Y.-L.; Li, G.-Q.; Yu, Q.; Yang, J. Identifications of Immune-Responsive Genes for Adaptative Traits by Comparative Transcriptome Analysis of Spleen Tissue from Kazakh and Suffolk Sheep. Sci. Rep. 2021, 11, 3157. [Google Scholar] [CrossRef]
- Wu, Q.; Han, Y.; Wu, X.; Wang, Y.; Su, Q.; Shen, Y.; Guan, K.; Michal, J.J.; Jiang, Z.; Liu, B.; et al. Integrated Time-Series Transcriptomic and Metabolomic Analyses Reveal Different Inflammatory and Adaptive Immune Responses Contributing to Host Resistance to PRRSV. Front. Immunol. 2022, 13, 960709. [Google Scholar] [CrossRef]
- Lopez, B.I.; Santiago, K.G.; Lee, D.; Ha, S.; Seo, K. RNA Sequencing (RNA-Seq) Based Transcriptome Analysis in Immune Response of Holstein Cattle to Killed Vaccine against Bovine Viral Diarrhea Virus Type I. Animals 2020, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Mao, L.; Shu, X.; Liu, R.; Jiang, J. Transcriptome Analysis Reveals Differential Immune Related Genes Expression in Bovine Viral Diarrhea Virus-2 Infected Goat Peripheral Blood Mononuclear Cells (PBMCs). BMC Genom. 2019, 20, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Fan, Y.; Yang, Z.; Loor, J.J.; Yang, Y. Mammary Transcriptome Profile during Peak and Late Lactation Reveals Differentially Expression Genes Related to Inflammation and Immunity in Chinese Holstein. Animals 2020, 10, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cassell, B.G. Sire-Daughter and Dam-Daughter Paths: Research Geneticists’ View. J. Dairy Sci. 1988, 71, 1993–2000. [Google Scholar] [CrossRef]
- Sire vs. Dam—Which Has a Greater Impact on Your Herd’s Genetic Improvement? The Bullvine—The Dairy Information You Want to Know When You Need It. Available online: https://www.thebullvine.com/dairy-cattle-reproduction/sire-vs-dam-greater-impact-herds-genetic-improvement/ (accessed on 28 February 2023).
- Larmer, S.G.; Mallard, B.A. High Immune Response Sires Reduce Disease Incidence in North American Large Commercial Dairy Populations. Cattle Pract. 2017, 25, 74–81. [Google Scholar]
- Liu, S.; Yue, T.; Ahmad, M.J.; Hu, X.; Zhang, X.; Deng, T.; Hu, Y.; He, C.; Zhou, Y.; Yang, L. Transcriptome Analysis Reveals Potential Regulatory Genes Related to Heat Tolerance in Holstein Dairy Cattle. Genes 2020, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Pawlik, A.; Sender, G.; Sobczyńska, M.; Korwin-Kossakowska, A.; Lassa, H.; Oprządek, J. Lactoferrin Gene Variants, Their Expression in the Udder and Mastitis Susceptibility in Dairy Cattle. Anim. Prod. Sci. 2015, 55, 999. [Google Scholar] [CrossRef]
- Raliou, M.; Dembélé, D.; Düvel, A.; Bolifraud, P.; Aubert, J.; Mary-Huard, T.; Rocha, D.; Piumi, F.; Mockly, S.; Heppelmann, M.; et al. Subclinical Endometritis in Dairy Cattle Is Associated with Distinct MRNA Expression Patterns in Blood and Endometrium. PLoS ONE 2019, 14, e0220244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.-E.; Shin, M.-K.; Park, H.-T.; Jung, M.; Cho, Y.I.; Yoo, H.S. Gene Expression Profiles of Putative Biomarker Candidates in Mycobacterium avium subsp. paratuberculosis-Infected Cattle. Pathog. Dis. 2016, 74, ftw022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tydell, C.C.; Yuan, J.; Tran, P.; Selsted, M.E. Bovine Peptidoglycan Recognition Protein-S: Antimicrobial Activity, Localization, Secretion, and Binding Properties. J. Immunol. 2006, 176, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.L.; Li, Z.X.; Wang, L.J.; He, H.; Yang, J.; Chen, L.; Niu, F.B.; Liu, Y.; Guo, J.Z.; Liu, X.L. Polymorphism in PGLYRP-1 Gene by PCR-RFLP and Its Association with Somatic Cell Score in Chinese Holstein. Res. Vet. Sci. 2013, 95, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Dinasarapu, A.; Isenman, D.; Subramaniam, S. Complement C5. UCSD Mol. Pages 2012, 1, 61–70. [Google Scholar] [CrossRef]
- Association between the SERPING1 Gene and Age-Related Macular Degeneration: A Two-Stage Case-Control Study|Request PDF. Available online: https://www.researchgate.net/publication/23306686_Association_between_the_SERPING1_gene_and_age-related_macular_degeneration_a_two-stage_case-control_study (accessed on 2 February 2023).
- Rennier, K.; Shin, W.; Krug, E.; Virdi, G.; Pachynski, R. Chemerin Reactivates PTEN and Suppresses PD-L1 in Tumor Cells via Modulation of a Novel CMKLR1-Mediated Signaling Cascade. Clin. Cancer Res. 2020, 26, 5019–5035. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.L.; Brodsky, I.E.; Mangalmurti, N.S. The Evolving Erythrocyte: Red Blood Cells as Modulators of Innate Immunity. J. Immunol. 2018, 201, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Coates, C.J.; Decker, H. Immunological Properties of Oxygen-Transport Proteins: Hemoglobin, Hemocyanin and Hemerythrin. Cell. Mol. Life Sci. 2017, 74, 293–317. [Google Scholar] [CrossRef] [Green Version]
- Nedjar-Arroume, N.; Dubois-Delval, V.; Miloudi, K.; Daoud, R.; Krier, F.; Kouach, M.; Briand, G.; Guillochon, D. Isolation and Characterization of Four Antibacterial Peptides from Bovine Hemoglobin. Peptides 2006, 27, 2082–2089. [Google Scholar] [CrossRef]
- Thompson-Crispi, K.A.; Sargolzaei, M.; Ventura, R.; Abo-Ismail, M.; Miglior, F.; Schenkel, F.; Mallard, B.A. A Genome-Wide Association Study of Immune Response Traits in Canadian Holstein Cattle. BMC Genom. 2014, 15, 559. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Luna, P.; Nag, R.; Martinez, I.; Mauricio-Iglesias, M.; Hospido, A.; Cummins, E. Quantifying Current and Future Raw Milk Losses Due to Bovine Mastitis on European Dairy Farms under Climate Change Scenarios. Sci. Total Environ. 2022, 833, 155149. [Google Scholar] [CrossRef] [PubMed]
- Molineri, A.I.; Camussone, C.; Zbrun, M.V.; Archilla, G.S.; Signorini, M. Antimicrobial Resistance of Staphylococcus Aureus Isolated from Bovine Mastitis: Systematic Review and Meta-Analysis. Prev. Vet. Med. 2021, 188, 105261. [Google Scholar] [CrossRef] [PubMed]
- Turrin, N.P.; Plata-Salamán, C.R. Cytokine-Cytokine Interactions and the Brain. Brain Res. Bull. 2000, 51, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Umeyama, Y.; Hama, N.; Kobayashi, T.; Han, N.; Wada, H.; Seino, K.-I. Interleukin-34, a Comprehensive Review. J. Leukoc. Biol. 2018, 104, 931–951. [Google Scholar] [CrossRef]
- Miles, A.M.; Huson, H.J. Time- and Population-Dependent Genetic Patterns Underlie Bovine Milk Somatic Cell Count. J. Dairy Sci. 2020, 103, 8292–8304. [Google Scholar] [CrossRef]
- Sharifi, S.; Pakdel, A.; Ebrahimi, M.; Reecy, J.M.; Fazeli Farsani, S.; Ebrahimie, E. Integration of Machine Learning and Meta-Analysis Identifies the Transcriptomic Bio-Signature of Mastitis Disease in Cattle. PLoS ONE 2018, 13, e0191227. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; van Eijk, M.J.; Park, C.; Lewin, H.A. Polymorphism in BoLA-DRB3 Exon 2 Correlates with Resistance to Persistent Lymphocytosis Caused by Bovine Leukemia Virus. J. Immunol. 1993, 151, 6977–6985. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Raw Reads | Clean Reads | Clean Bases | Error Rate (%) | Q30 (%) | GC Content (%) | Uniquely Mapping Rate (%) |
|---|---|---|---|---|---|---|---|
| H1 | 46,711,716 | 46,104,046 | 6.18 G | 0.03 | 94.26 | 52.74 | 92.96% |
| H2 | 41,180,040 | 40,655,298 | 5.45 G | 0.03 | 93.87 | 52.40 | 92.71% |
| H3 | 45,339,678 | 44,767,294 | 6.0 G | 0.03 | 93.80 | 52.73 | 93.12% |
| L1 | 41,720,406 | 41,196,110 | 5.51 G | 0.03 | 94.38 | 53.22 | 89.97% |
| L2 | 47,547,798 | 46,913,208 | 6.29 G | 0.03 | 93.45 | 52.57 | 92.52% |
| Pathway | ID | p-Value | Upregulated Genes | Downregulated Genes |
|---|---|---|---|---|
| Graft-versus-host disease | bta05332 | 6.14 × 10−5 | BOLA-DQB, BOLA-DQA5, KIR3DL1, ENSBTAG00000000966, ENSBTAG00000049367, ENSBTAG00000052514 | JSP.1, KIR3DL2, ENSBTAG00000039813, ENSBTAG00000049260 |
| ECM–receptor interaction | bta04512 | 0.000177 | COL1A1, COL1A2, COL6A1, COL6A2, GP5, LAMA4, LAMC2, SV2C | ITGA9, ITGB4, TNR |
| Antigen processing and presentation | bta04612 | 0.000895 | ENSBTAG00000000966, ENSBTAG00000049367, ENSBTAG00000052514, BOLA-DQA5, BOLA-DQB, CTSV, KIR3DL1 | ENSBTAG00000049260, JSP.1, KIR3DL2 |
| Systemic lupus erythematosus | bta05322 | 0.001285 | ENSBTAG00000006864, ENSBTAG00000048268, BOLA-DQA5, BOLA-DQB, C5, H2AC8 | ENSBTAG00000038433, ENSBTAG00000049260, H2BC13, H2BU1 |
| Staphylococcus aureus infection | bta05150 | 0.003058 | ENSBTAG00000006864, ENSBTAG00000048268, BOLA-DQA5, BOLA-DQB, C5, FGG, KRT18, KRT26 | ENSBTAG00000049260, KRT23 |
| Intestinal immune network for IgA production | bta04672 | 0.003073 | ENSBTAG00000048268, BOLA-DQA5, BOLA-QB, CCR10, CXCL12, TNFRSF17 | ENSBTAG00000049260 |
| Viral myocarditis | bta05416 | 0.004436 | MYH7, BOLA-DQB, BOLA-DQA5, MYH6, ENSBTAG00000048268 | JSP.1, CYCT, ENSBTAG00000049260 |
| Cytokine–cytokine receptor interaction | bta04060 | 0.008272 | CCR10, CX3CL1, CXCL12, GH1, IL1RL2, IL34, IL36A, INHA, MPL, TNFRSF17, TNFSF15, ENSBTAG00000052397 | BMP6, CCL19, CXCL2, GDF9, GRO1, IL5RA, TGFB2, TNFSF4 |
| Autoimmune thyroid disease | bta05320 | 0.010892 | TSHB, BOLA-DQB, BOLA-DQA5, ENSBTAG00000048268 | JSP.1, ENSBTAG00000039813, ENSBTAG00000049260 |
| Complement and coagulation cascades | bta04610 | 0.01253 | C5, F2, F2RL3, FGG, SERPING1, ENSBTAG00000006864, ENSBTAG00000023026 | PROS1 |
| Allograft rejection | bta05330 | 0.012633 | BOLA-DQB, BOLA-DQA5, ENSBTAG00000048268 | JSP.1, ENSBTAG00000039813, ENSBTAG00000049260 |
| TGF-beta signaling pathway | bta04350 | 0.036167 | ENSBTAG00000005140, DCN | FBN1, BAMBI, TGFB2, HAMP, BMP6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Luo, H.; Lu, H.; Ma, L.; Li, Y.; Dou, J.; Zhang, J.; Ma, Y.; Li, J.; Wang, Y. RNA-Seq Analysis of Peripheral Whole Blood from Dairy Bulls with High and Low Antibody-Mediated Immune Responses—A Preliminary Study. Animals 2023, 13, 2208. https://doi.org/10.3390/ani13132208
Zhao X, Luo H, Lu H, Ma L, Li Y, Dou J, Zhang J, Ma Y, Li J, Wang Y. RNA-Seq Analysis of Peripheral Whole Blood from Dairy Bulls with High and Low Antibody-Mediated Immune Responses—A Preliminary Study. Animals. 2023; 13(13):2208. https://doi.org/10.3390/ani13132208
Chicago/Turabian StyleZhao, Xiuxin, Hanpeng Luo, Haibo Lu, Longgang Ma, Yanqin Li, Jinhuan Dou, Junxing Zhang, Yun Ma, Jianbin Li, and Yachun Wang. 2023. "RNA-Seq Analysis of Peripheral Whole Blood from Dairy Bulls with High and Low Antibody-Mediated Immune Responses—A Preliminary Study" Animals 13, no. 13: 2208. https://doi.org/10.3390/ani13132208
APA StyleZhao, X., Luo, H., Lu, H., Ma, L., Li, Y., Dou, J., Zhang, J., Ma, Y., Li, J., & Wang, Y. (2023). RNA-Seq Analysis of Peripheral Whole Blood from Dairy Bulls with High and Low Antibody-Mediated Immune Responses—A Preliminary Study. Animals, 13(13), 2208. https://doi.org/10.3390/ani13132208