
A Nonsense Variant in the DMD Gene Causes X-Linked Muscular Dystrophy in the Maine Coon Cat
 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Examination
2.2. Sample Collection
2.3. Histopathology and Immunofluorescent Staining
2.4. Muscle mRNA
2.4.1. mRNA Expression
2.4.2. Variant Identification
2.5. Variant Confirmation and MYBPC3 Genotyping
2.6. Population Screening
2.7. Variant Classification
3. Results
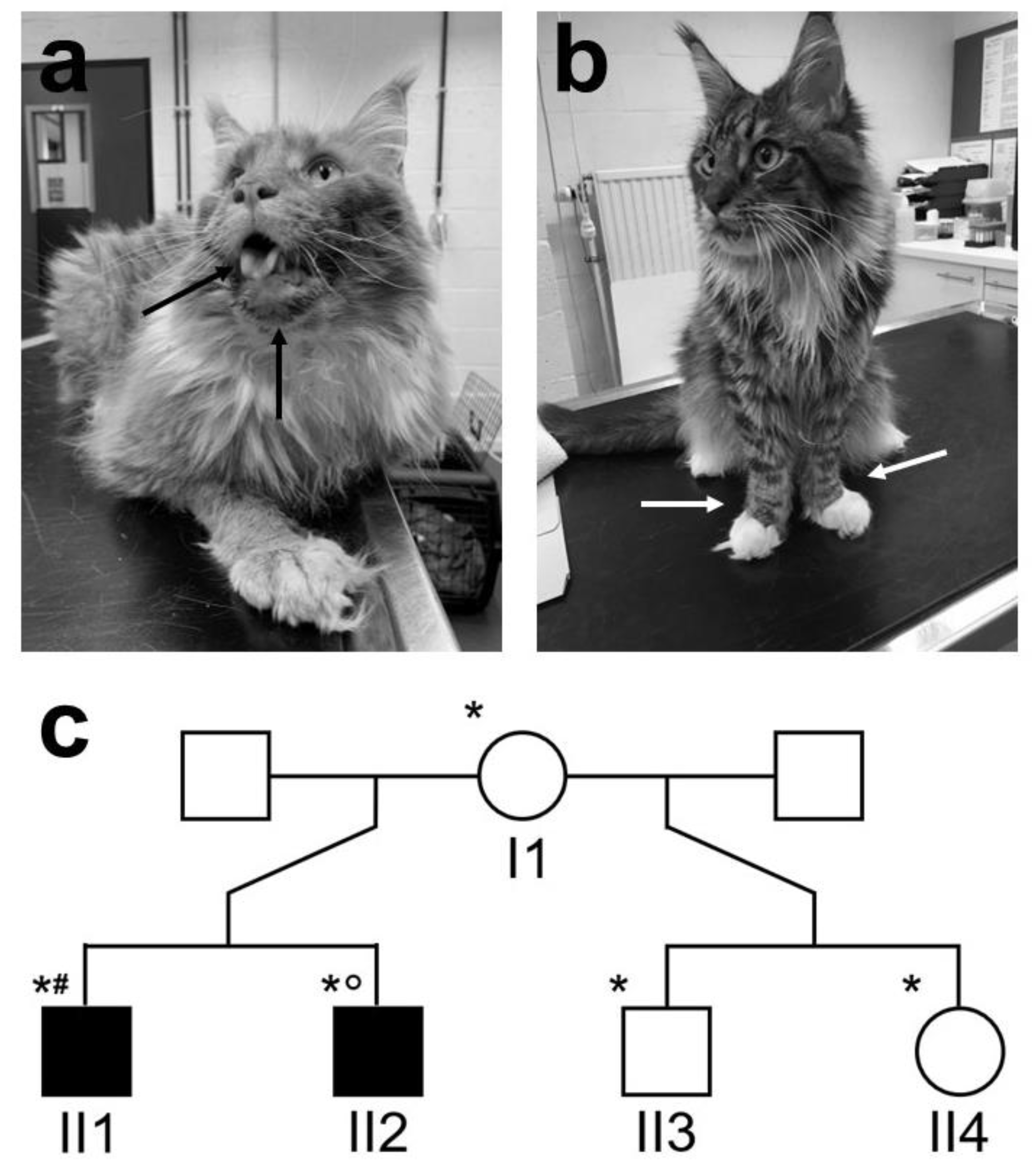
3.1. Clinical Findings
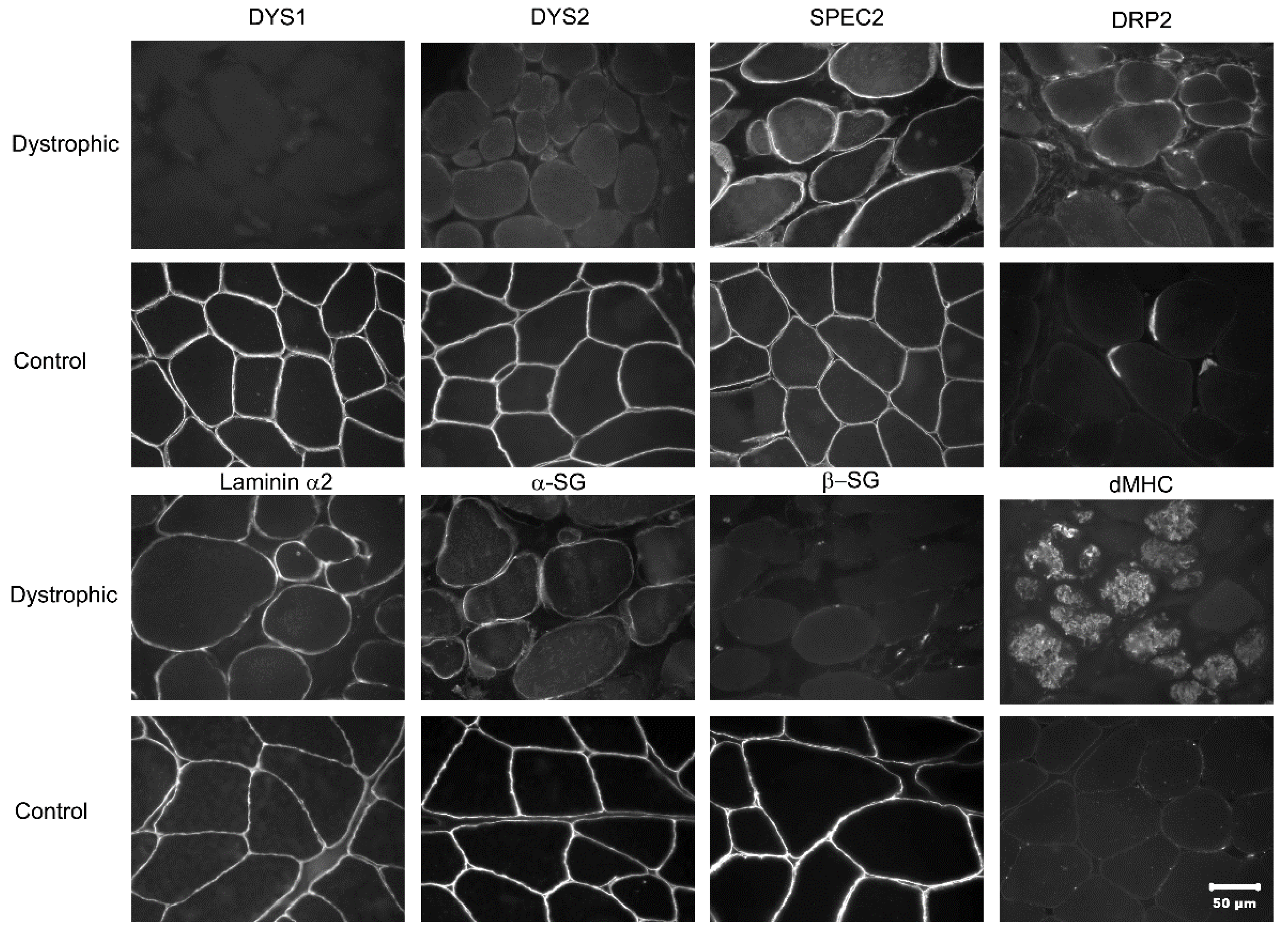
3.2. Histopathology and Immunofluorescent Staining
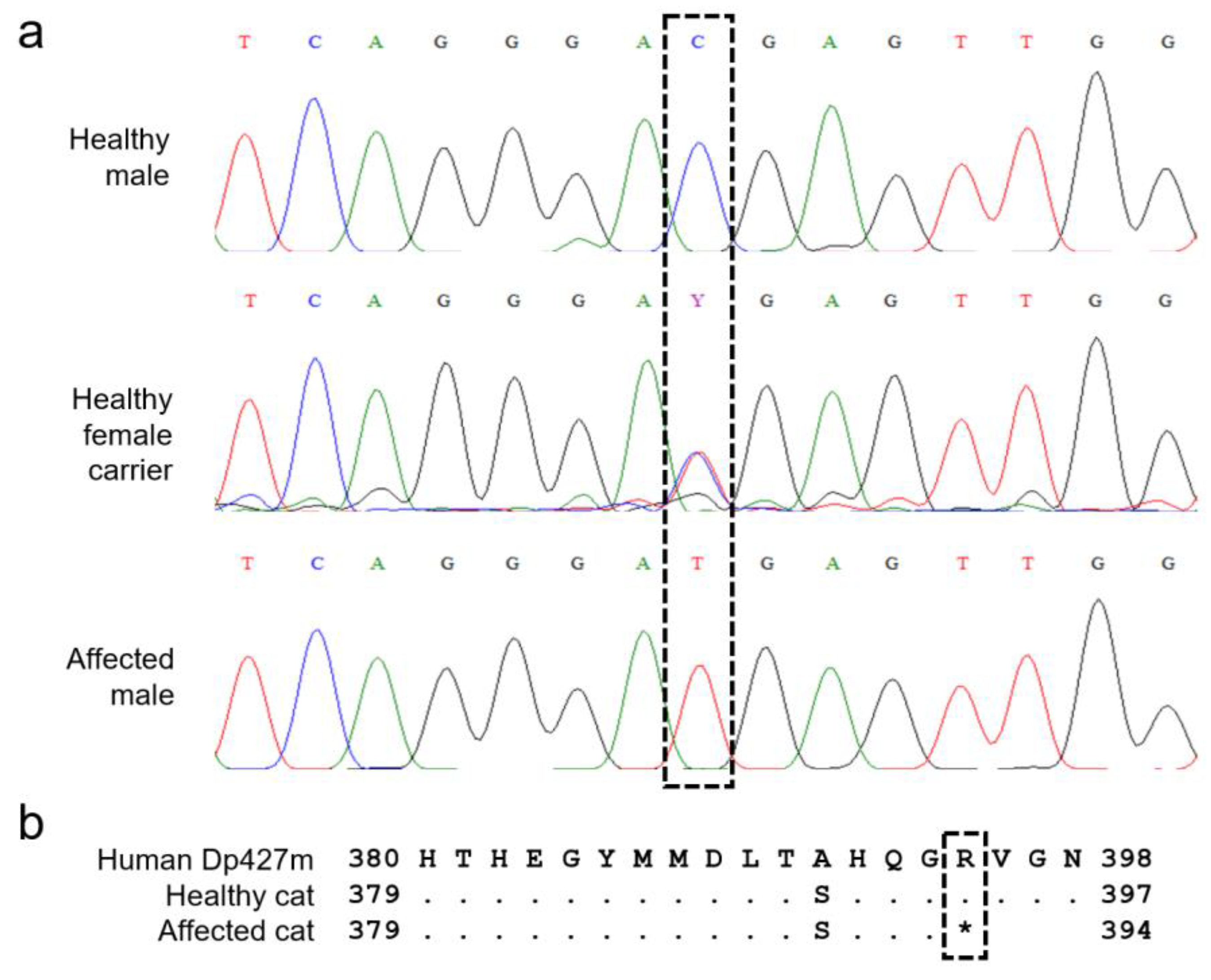
3.3. Muscle mRNA Expression and Variant Identification
3.4. Variant Confirmation, Population Screening, and MYBPC3 Genotyping
3.5. ACMG Classification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; De Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Douglas, A.G.; Wood, M.J. Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 2013, 56, 169–185. [Google Scholar] [CrossRef]
- Kong, X.; Zhong, X.; Liu, L.; Cui, S.; Yang, Y.; Kong, L. Genetic analysis of 1051 Chinese families with Duchenne/Becker Muscular Dystrophy. BMC Med. Genet. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Yucel, N.; Chang, A.C.; Day, J.W.; Rosenthal, N.; Blau, H.M. Humanizing the mdx mouse model of DMD: The long and the short of it. NPJ Regen. Med. 2018, 3, 4. [Google Scholar] [CrossRef]
- Taglietti, V.; Kefi, K.; Bronisz-Budzyńska, I.; Mirciloglu, B.; Rodrigues, M.; Cardone, N.; Coulpier, F.; Periou, B.; Gentil, C.; Goddard, M.; et al. Duchenne muscular dystrophy trajectory in R-DMDdel52 preclinical rat model identifies COMP as biomarker of fibrosis. Acta Neuropathol. Commun. 2022, 10, 60. [Google Scholar] [CrossRef]
- Sui, T.; Lau, Y.S.; Liu, D.; Liu, T.; Xu, L.; Gao, Y.; Lai, L.; Li, Z.; Han, R. A novel rabbit model of Duchenne muscular dystrophy generated by CRISPR/Cas9. Dis. Model. Mech. 2018, 11, dmm032201. [Google Scholar] [CrossRef]
- Hollinger, K.; Yang, C.X.; Montz, R.E.; Nonneman, D.; Ross, J.W.; Selsby, J.T. Dystrophin insufficiency causes selective muscle histopathology and loss of dystrophin-glycoprotein complex assembly in pig skeletal muscle. FASEB J. 2013, 28, 1600–1609. [Google Scholar] [CrossRef]
- Barthélémy, I.; Calmels, N.; Weiss, R.B.; Tiret, L.; Vulin, A.; Wein, N.; Peccate, C.; Drougard, C.; Beroud, C.; Deburgrave, N.; et al. X-linked muscular dystrophy in a Labrador Retriever strain: Phenotypic and molecular characterisation. Skelet. Muscle 2020, 10, 23. [Google Scholar] [CrossRef]
- Brunetti, B.; Muscatello, L.V.; Letko, A.; Papa, V.; Cenacchi, G.; Grillini, M.; Murgiano, L.; Jagannathan, V.; Drögemüller, C. X-Linked Duchenne-Type Muscular Dystrophy in Jack Russell Terrier Associated with a Partial Deletion of the Canine DMD Gene. Genes 2020, 11, 1175. [Google Scholar] [CrossRef] [PubMed]
- Shelton, G.D.; Minor, K.M.; Vieira, N.M.; Kunkel, L.M.; Friedenberg, S.G.; Cullen, J.N.; Guo, L.T.; Zatz, M.; Mickelson, J.R. Tandem Duplication within the DMD gene in Labrador Retrievers with a Mild Clinical Phenotype. Neuromuscul. Disord. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Gambino, A.N.; Mouser, P.J.; Shelton, G.D.; Winand, N.J. Emergent Presentation of a Cat with Dystrophin-Deficient Muscular Dystrophy. J. Am. Anim. Hosp. Assoc. 2014, 50, 130–135. [Google Scholar] [CrossRef]
- Winand, N.J.; Edwards, M.; Pradhan, D.; Berian, C.A.; Cooper, B.J. Deletion of the dystrophin muscle promotor in feline muscular dystrophy. Neuromuscul. Disord. 1994, 4, 433–445. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Y.; Kang, Y.; Yang, W.; Niu, Y.; Guo, X.; Tu, Z.; Si, C.; Wang, H.; Xing, R.; et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 2015, 24, 3764–3774. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Spurney, C.F.; Nghiem, P.P.; Brinkmeyer-Langford, C.L.; Hoffman, E.; Nagaraju, K. Pharmacologic Management of Duchenne Muscular Dystrophy: Target Identification and Preclinical Trials. ILAR J. 2014, 55, 119–149. [Google Scholar] [CrossRef]
- Kornegay, J.N.; Bogan, J.R.; Bogan, D.J.; Childers, M.; Li, J.; Nghiem, P.; Detwiler, D.A.; Larsen, C.A.; Grange, R.W.; Bhavaraju-Sanka, R.K.; et al. Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm. Genome 2012, 23, 85–108. [Google Scholar] [CrossRef]
- Marden, F.A.; Connolly, A.M.; Siegel, M.J.; Rubin, D. Compositional analysis of muscle in boys with Duchenne muscular dystrophy using MR imaging. Skelet. Radiol. 2004, 34, 140–148. [Google Scholar] [CrossRef]
- Shieh, P.B. Muscular Dystrophies and Other Genetic Myopathies. Neurol. Clin. 2013, 31, 1009–1029. [Google Scholar] [CrossRef]
- Dickinson, P.J.; LeCouteur, R.A. Muscle and nerve biopsy. Vet. Clin. N. Am. Small Anim. Pract. 2002, 32, 63–102. [Google Scholar] [CrossRef]
- Dubowitz, V.; Sewry, C.; Oldfors, A. Muscle Biopsy: A practical approach. In Muscle Biopsy: A Practical Approach, 4th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2013; pp. 16–27. [Google Scholar] [CrossRef]
- Van Poucke, M.; Martlé, V.; Van Brantegem, L.; Ducatelle, R.; Van Ham, L.; Bhatti, S.; Peelman, L.J. A canine orthologue of the human GFAP c.716G>A (p.Arg239His) variant causes Alexander disease in a Labrador retriever. Eur. J. Hum. Genet. 2015, 24, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Van Poucke, M.; Vandesompele, J.; Mattheeuws, M.; Van Zeveren, A.; Peelman, L.J. A dual fluorescent multiprobe assay for prion protein genotyping in sheep. BMC Infect. Dis. 2005, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Pagel, K.A.; Pejaver, V.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; Mooney, S.D.; Radivojac, P. When loss-of-function is loss of function: Assessing mutational signatures and impact of loss-of-function genetic variants. Bioinformatics 2017, 33, i389–i398. [Google Scholar] [CrossRef]
- Meurs, K.M.; Sanchez, X.; David, R.M.; Bowles, N.E.; Towbin, J.A.; Reiser, P.J.; Kittleson, J.A.; Munro, M.J.; Dryburgh, K.; Macdonald, K.A.; et al. A cardiac myosin binding protein C mutation in the Maine Coon cat with familial hypertrophic cardiomyopathy. Hum. Mol. Genet. 2005, 14, 3587–3593. [Google Scholar] [CrossRef]
- Lyons, L.A.; Buckley, R.M.; Harvey, R.J.; The 99 Lives Cat Genome Consortium; Abitbol, M.; Aberdein, D.; Alves, P.C.; Andersson, A.O.; Bellone, R.R.; Bergström, T.F.; et al. Mining the 99 Lives Cat Genome Sequencing Consortium database implicates genes and variants for the Ticked locus in domestic cats (Felis catus). Anim. Genet. 2021, 52, 321–332. [Google Scholar] [CrossRef]
- Cezard, T.; Cunningham, F.; Hunt, S.E.; Koylass, B.; Kumar, N.; Saunders, G.; Shen, A.; Silva, A.F.; Tsukanov, K.; Venkataraman, S.; et al. The European Variation Archive: A FAIR resource of genomic variation for all species. Nucleic Acids Res. 2021, 50, D1216–D1220. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kube, S.A.; Vernau, K.M.; LeCouteur, R.A.; Mizisin, A.P.; Shelton, G.D. Congenital myopathy with abundant nemaline rods in a cat. Neuromuscul. Disord. 2006, 16, 188–191. [Google Scholar] [CrossRef]
- Kopke, M.A.; Shelton, G.D.; Lyons, L.A.; Wall, M.J.; Pemberton, S.; Gedye, K.R.; Owen, R.; Guo, L.T.; Buckley, R.M.; Valencia, J.A.; et al. X-linked myotubular myopathy associated with an MTM1 variant in a Maine coon cat. J. Vet. Intern. Med. 2022, 26, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Poncelet, L.; Résibois, A.; Engvall, E.; Shelton, G.D. Laminin alpha2 deficiency-associated muscular dystrophy in a Maine coon cat. Proc. Natl. Acad. Sci. USA 2003, 44, 550–552. [Google Scholar]
- Bouillon, J.; Taylor, S.M.; Vargo, C.; Lange, M.; Zwicker, L.A.; Sukut, S.L.; Guo, L.T.; Shelton, G.D. Beta-sarcoglycan-deficient muscular dystrophy presenting as chronic bronchopneumonia in a young cat. J. Feline Med. Surg. Open Rep. 2019, 5, 2055116919856457. [Google Scholar] [CrossRef] [PubMed]
- Gaschen, L.; Lang, J.; Lin, S.; Adé-Damilano, M.; Busato, A.; Lombard, C.W.; Gaschen, F.P. Cardiomyopathy in Dystrophin-Deficient Hypertrophic Feline Muscular Dystrophy. J. Vet. Intern. Med. 1999, 13, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Longeri, M.; Ferrari, P.; Knafelz, P.; Mezzelani, A.; Marabotti, A.; Milanesi, L.; Pertica, G.; Polli, M.; Brambilla, P.; Kittleson, M.; et al. Myosin-Binding Protein C DNA Variants in Domestic Cats (A31P, A74T, R820W) and their Association with Hypertrophic Cardiomyopathy. J. Vet. Intern. Med. 2013, 27, 275–285. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Sheri, N.; Nguyen, Q.; Yokota, T. Cardiac Involvement in Dystrophin-Deficient Females: Current Understanding and Implications for the Treatment of Dystrophinopathies. Genes 2020, 11, 765. [Google Scholar] [CrossRef]
- Zhang, J.; Meng, Q.; Zhong, J.; Zhang, M.; Qin, X.; Ni, X.; Ma, J.; He, Y.; Zeng, D.; Lan, D. Serum MyomiRs as Biomarkers for Female Carriers of Duchenne/Becker Muscular Dystrophy. Front. Neurol. 2020, 11, 563609. [Google Scholar] [CrossRef]
- Howard, J.; Jaggy, A.; Busato, A.; Gaschen, F. Electrodiagnostic evaluation in feline hypertrophic muscular dystrophy. Vet. J. 2004, 168, 87–92. [Google Scholar] [CrossRef]
- Haslett, J.N.; Sanoudou, D.; Kho, A.T.; Bennett, R.R.; Greenberg, S.A.; Kohane, I.S.; Beggs, A.H.; Kunkel, L.M. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 15000–15005. [Google Scholar] [CrossRef]
- Chamberlain, J.S.; Pearlman, J.A.; Muzny, D.M.; Gibbs, R.A.; Ranier, J.E.; Reeves, A.A.; Caskey, C.T. Expression of the Murine Duchenne Muscular Dystrophy Gene in Muscle and Brain. Science 1988, 239, 1416–1418. [Google Scholar] [CrossRef]
- Nghiem, P.P.; Bello, L.; Balog-Alvarez, C.; López, S.M.; Bettis, A.; Barnett, H.; Hernandez, B.; Schatzberg, S.J.; Piercy, R.J.; Kornegay, J.N. Whole genome sequencing reveals a 7 base-pair deletion in DMD exon 42 in a dog with muscular dystrophy. Mamm. Genome 2016, 28, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Vogel, H.; Zamecnik, J. Diagnostic Immunohistology of Muscle Diseases. J. Neuropathol. Exp. Neurol. 2005, 64, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The Molecular Basis of Muscular Dystrophy in the mdx Mouse: A Point Mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Torella, A.; Zanobio, M.; Zeuli, R.; Del Vecchio Blanco, F.; Savarese, M.; Giugliano, T.; Garofalo, A.; Piluso, G.; Politano, L.; Nigro, V. The position of nonsense mutations can predict the phenotype severity: A survey on the DMD gene. PLoS ONE 2020, 15, e0237803. [Google Scholar] [CrossRef]
- Doorenweerd, N.; Mahfouz, A.; Van Putten, M.; Kaliyaperumal, R.; Hoen, P.A.; Hendriksen, J.G.M.; Aartsma-Rus, A.; Verschuuren, J.J.G.M.; Niks, E.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef]
- Liu, L.A.; Engvall, E. Sarcoglycan Isoforms in Skeletal Muscle. J. Biol. Chem. 1999, 274, 38171–38176. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Criterion | Result | Remarks | Conclusion |
|---|---|---|---|
| Null variant in a gene where LOF is a known mechanism of disease (PVS1) | LOF confirmed at protein level | Presence of alternate gene transcripts, but the variant affects the biologically relevant transcript | PVS1 fulfilled |
| Functional studies show deleterious (PS3) or no deleterious (BS3) effect | Abundance of functional studies of the human DMD gene show a deleterious effect | PS3 fulfilled | |
| Significant OR > 5 (PS4) | OR = infinite; p = 0.02 | CI could not be calculated because of perfect segregation, so p value was calculated | PS4 fulfilled |
| Variant absent in population databases (PM2) | Variant absent in two databases and a Belgian Maine coon cohort | PM2 fulfilled | |
| Co-segregation in multiple affected family members (PP1) | Perfect variant segregation with FXMD phenotype in the pedigree | Limited sample availability, but segregation confirmed in two affected members | PP1 fulfilled |
| Multiple lines of computational evidence (PP3) | 2/2 programs predicted deleterious/pathogenic effects | PP3 fulfilled |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckers, E.; Cornelis, I.; Bhatti, S.F.M.; Smets, P.; Shelton, G.D.; Guo, L.T.; Peelman, L.; Broeckx, B.J.G. A Nonsense Variant in the DMD Gene Causes X-Linked Muscular Dystrophy in the Maine Coon Cat. Animals 2022, 12, 2928. https://doi.org/10.3390/ani12212928
Beckers E, Cornelis I, Bhatti SFM, Smets P, Shelton GD, Guo LT, Peelman L, Broeckx BJG. A Nonsense Variant in the DMD Gene Causes X-Linked Muscular Dystrophy in the Maine Coon Cat. Animals. 2022; 12(21):2928. https://doi.org/10.3390/ani12212928
Chicago/Turabian StyleBeckers, Evy, Ine Cornelis, Sofie F. M. Bhatti, Pascale Smets, G. Diane Shelton, Ling T. Guo, Luc Peelman, and Bart J. G. Broeckx. 2022. "A Nonsense Variant in the DMD Gene Causes X-Linked Muscular Dystrophy in the Maine Coon Cat" Animals 12, no. 21: 2928. https://doi.org/10.3390/ani12212928
APA StyleBeckers, E., Cornelis, I., Bhatti, S. F. M., Smets, P., Shelton, G. D., Guo, L. T., Peelman, L., & Broeckx, B. J. G. (2022). A Nonsense Variant in the DMD Gene Causes X-Linked Muscular Dystrophy in the Maine Coon Cat. Animals, 12(21), 2928. https://doi.org/10.3390/ani12212928