
Host Species Affects Bacterial Evenness, but Not Diversity: Comparison of Fecal Bacteria of Cows and Goats Offered the Same Diet

 ,
,  , , , , and
, , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. DNA Extraction
2.3. PCR Amplification and Purification
2.4. Library Perpetration and High-Throughput Sequencing
2.5. Bioinformatic Analysis
2.6. Statistical Analysis
3. Results
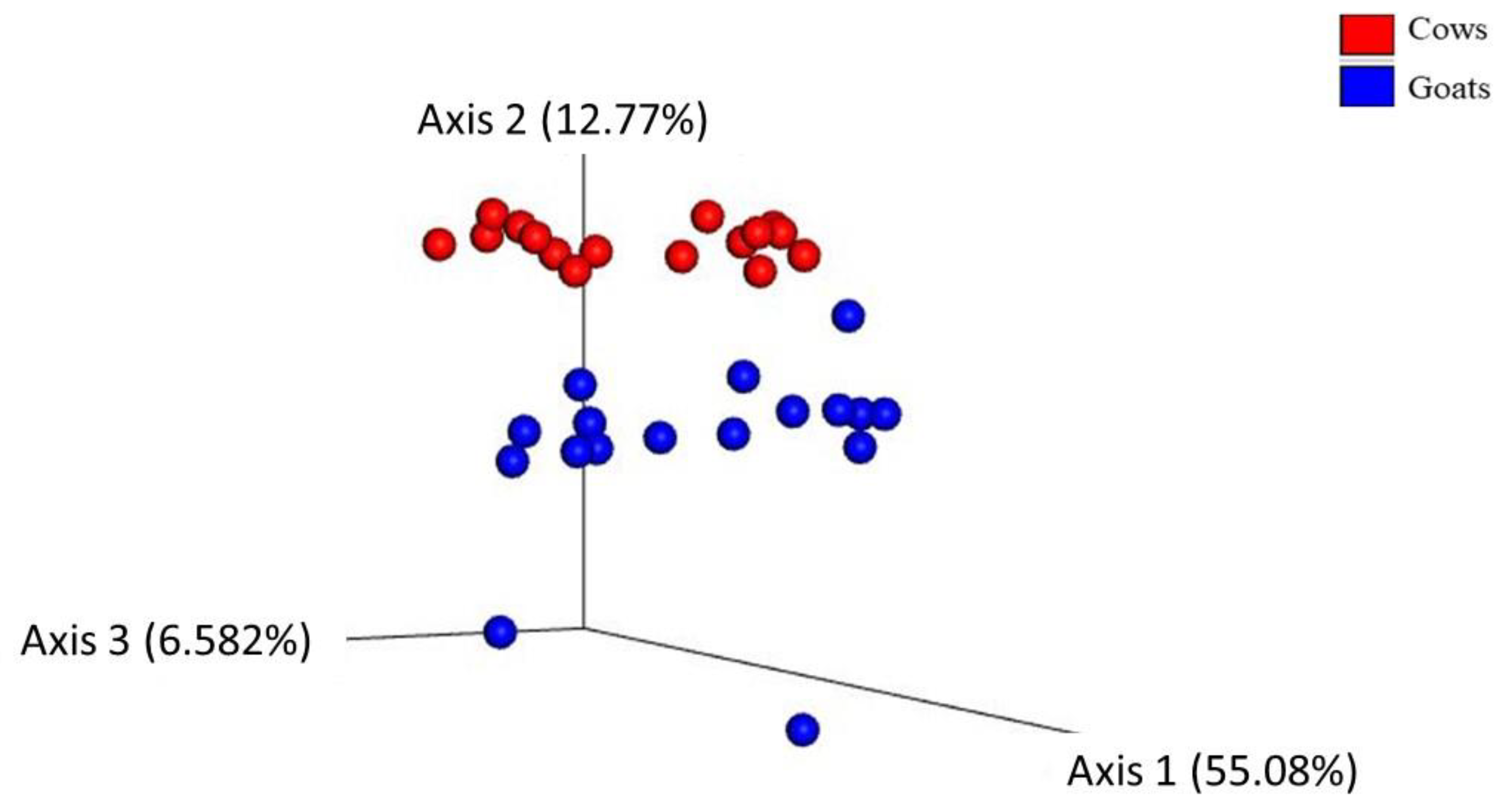
3.1. Alpha and Beta Diversity
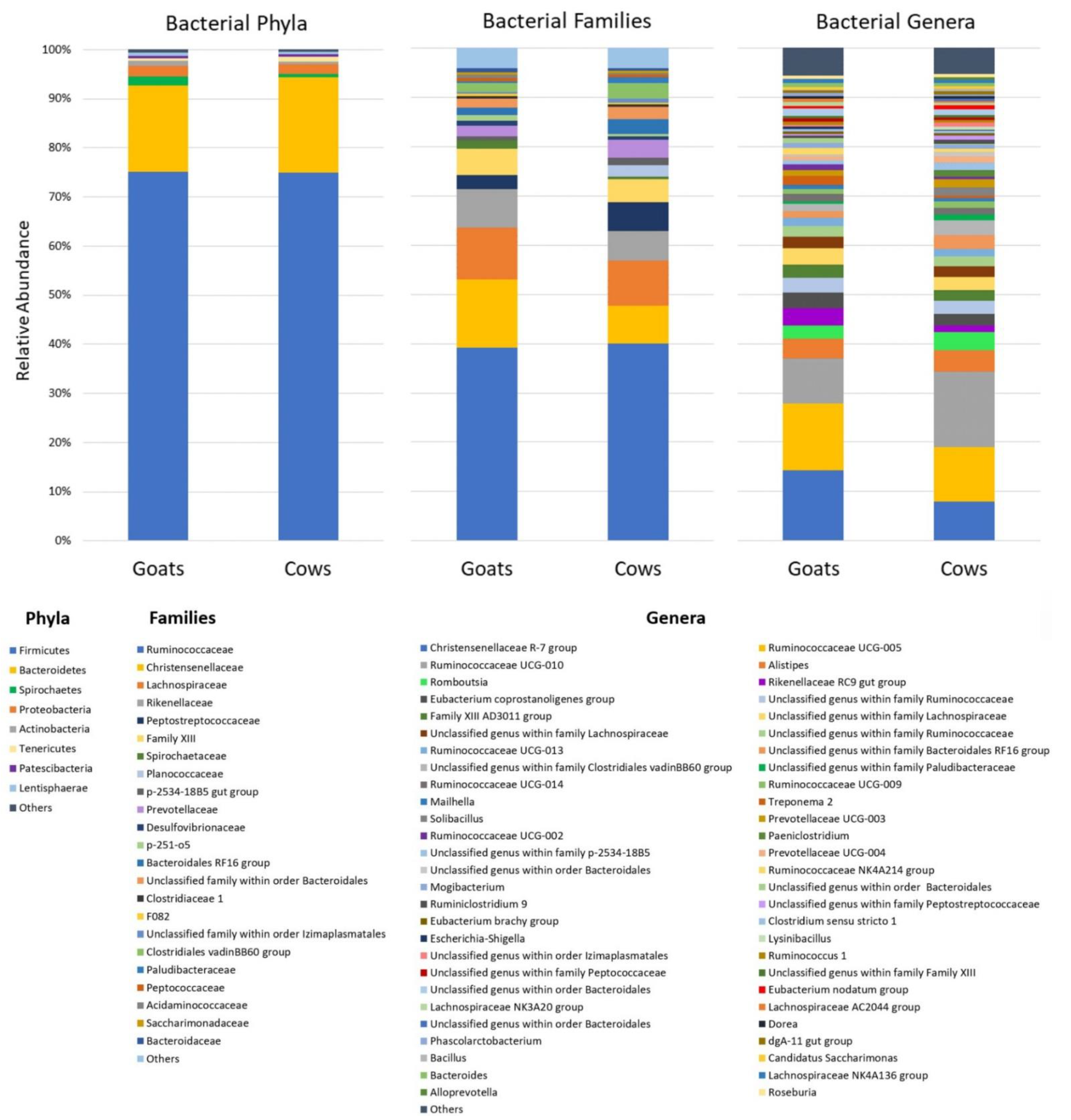
3.2. Taxonomical Composition
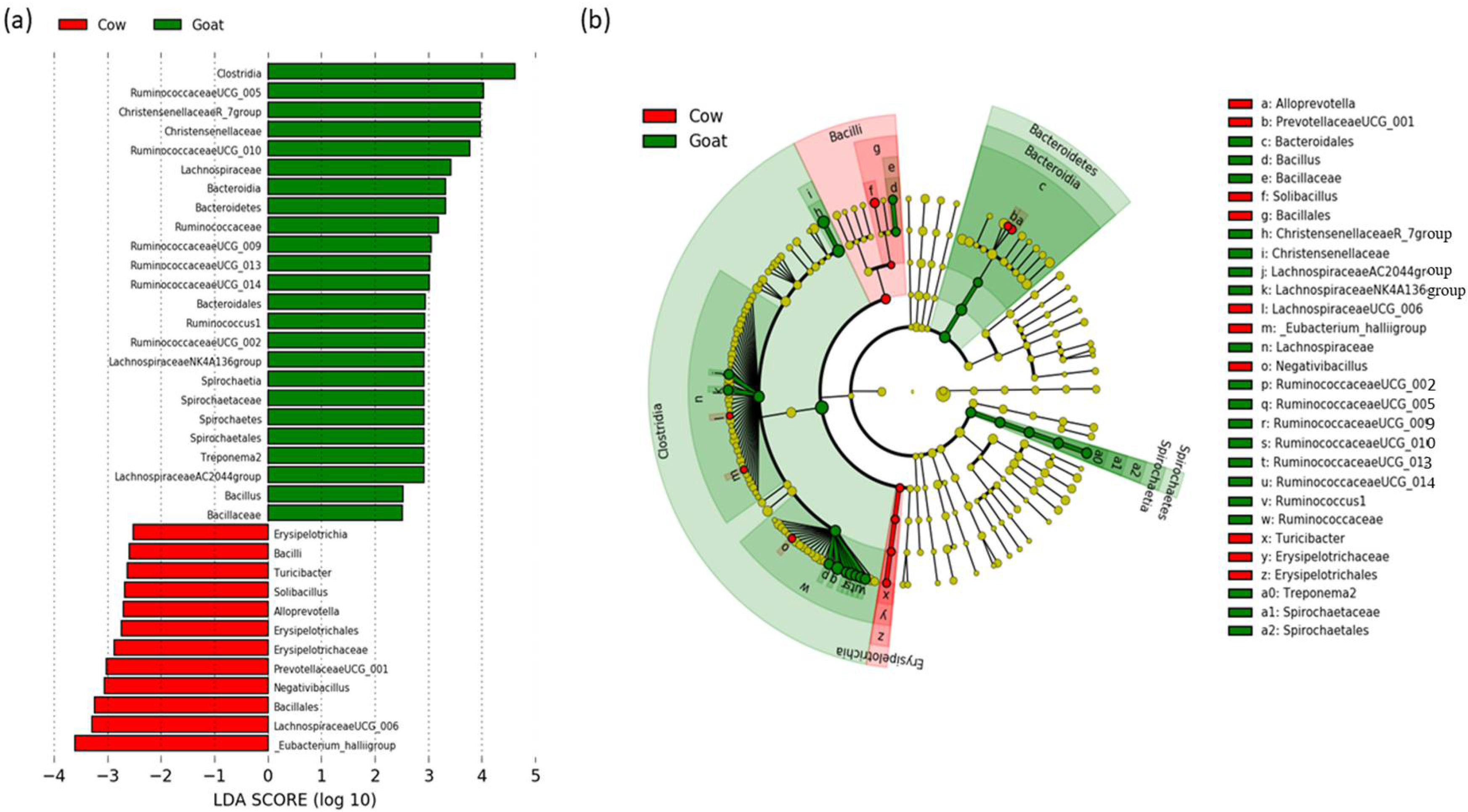
3.3. Determination of Taxonomic Biomarkers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abubakar, M.; Iqbal, A.; Kabir, A.; Manzoor, S. Introductory Chapter: Ruminants—The Husbandry, Economic, and Health Aspects. In Ruminants-The Husbandry, Economic and Health Aspects; IntechOpen: London, UK, 2018; pp. 3–8. [Google Scholar] [CrossRef][Green Version]
- Pulina, G.; Francesconi, A.H.D.; Stefanon, B.; Sevi, A.; Calamari, L.; Lacetera, N.; Dell’Orto, V.; Pilla, F.; Marsan, P.A.; Mele, M.; et al. Sustainable ruminant production to help feed the planet. Ital. J. Anim. Sci. 2017, 16, 140–171. [Google Scholar] [CrossRef]
- Hodgson, H.J. Role of the Dairy Cow in World Food Production. J. Dairy Sci. 1979, 62, 343–351. [Google Scholar] [CrossRef]
- Mazinani, M.; Rude, B. Population, world production and quality of sheep and goat products. Am. J. Anim. Vet. Sci. 2020, 15, 291–299. [Google Scholar] [CrossRef]
- Huws, S.A.; Creevey, C.J.; Oyama, L.B.; Mizrahi, I.; Denman, S.E.; Popova, M.; Muñoz-Tamayo, R.; Forano, E.; Waters, S.M.; Hess, M.; et al. Addressing global ruminant agricultural challenges through understanding the rumen microbiome: Past, present, and future. Front. Microbiol. 2018, 9, 1–33. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Li, Y.; Zhang, Y. Effects of high forage/concentrate diet on volatile fatty acid production and the microorganisms involved in VFA production in cow rumen. Animals 2020, 10, 223. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H.; Abecia, L.; Angarita, E.; Aravena, P.; Arenas, G.N.; et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- Fliegerova, K.O.; Podmirseg, S.M.; Vinzelj, J.; Grilli, D.J.; Kvasnová, S.; Schierová, D.; Sechovcová, H.; Mrázek, J.; Siddi, G.; Arenas, G.N.; et al. The effect of a high-grain diet on the rumen microbiome of goats with a special focus on anaerobic fungi. Microorganisms 2021, 9, 157. [Google Scholar] [CrossRef]
- Hua, C.; Tian, J.; Tian, P.; Cong, R.; Luo, Y.; Geng, Y.; Tao, S.; Ni, Y.; Zhao, R. Feeding a high concentration diet induces unhealthy alterations in the composition and metabolism of ruminal microbiota and host response in a goat model. Front. Microbiol. 2017, 8, 138. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Liu, Y.J.; Yin, Y.Y.; Jin, W.; Mao, S.Y.; Liu, J.H. Response of rumen microbiota, and metabolic profiles of rumen fluid, liver and serum of goats to high-grain diets. Animal 2019, 13, 1855–1864. [Google Scholar] [CrossRef]
- Plaizier, J.C.; Li, S.; Tun, H.M.; Khafipour, E. Nutritional models of experimentally-induced subacute ruminal acidosis (SARA) differ in their impact on rumen and hindgut bacterial communities in dairy cows. Front. Microbiol. 2017, 7, 2128. [Google Scholar] [CrossRef]
- Grilli, D.J.; Fliegerová, K.; Kopečný, J.; Lama, S.P.; Egea, V.; Sohaefer, N.; Pereyra, C.; Ruiz, M.S.; Sosa, M.A.; Arenas, G.N.; et al. Analysis of the rumen bacterial diversity of goats during shift from forage to concentrate diet. Anaerobe 2016, 42, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.Y.; Huo, W.J.; Zhu, W.Y. Microbiome-metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ. Microbiol. 2016, 18, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.M.; Hervás, G.; Belenguer, A.; Celaya, R.; Rodrigues, M.A.M.; García, U.; Frutos, P.; Osoro, K. Comparison of feed intake, digestion and rumen function among domestic ruminant species grazing in upland vegetation communities. J. Anim. Physiol. Anim. Nutr. 2017, 101, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Li, Z.; Ao, W.; Zhao, G.; Li, G.; Wu, J. Bacterial community composition and fermentation in the rumen of Xinjiang brown cattle (Bos taurus), Tarim red deer (Cervus elaphus yarkandensis), and Karakul sheep (Ovis aries). Can. J. Microbiol. 2017, 63, 375–383. [Google Scholar] [CrossRef]
- Zhang, T.; Mu, Y.; Zhang, D.; Lin, X.; Wang, Z.; Hou, Q.; Wang, Y.; Hu, Z. Determination of microbiological characteristics in the digestive tract of different ruminant species. Microbiologyopen 2019, 8, e00769. [Google Scholar] [CrossRef]
- Zhang, K.; Li, B.; Guo, M.; Liu, G.; Yang, Y.; Wang, X.; Chen, Y.; Zhang, E. Maturation of the goat rumen microbiota involves three stages of microbial colonization. Animals 2019, 9, 1028. [Google Scholar] [CrossRef]
- Fonty, G.; Gouet, P.; Jouany, J.; Senaud, J. Establishment of the Microflora and Anaerobic Fungi in the Rumen of Lambs. J. Gen. Microbiol. 1987, 133, 1835–1836. [Google Scholar] [CrossRef]
- Dias, J.; Marcondes, M.I.; Noronha, M.F.; Resende, R.T.; Machado, F.S.; Mantovani, H.C.; Dill-McFarland, K.A.; Suen, G. Effect of pre-weaning diet on the ruminal archaeal, bacterial, and fungal communities of dairy calves. Front. Microbiol. 2017, 8, 1553. [Google Scholar] [CrossRef]
- Liu, C.; Meng, Q.; Chen, Y.; Xu, M.; Shen, M.; Gao, R.; Gan, S. Role of age-related shifts in rumen bacteria and methanogens in methane production in cattle. Front. Microbiol. 2017, 8, 1563. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, D.; Wang, L.; Hao, J.; Wang, J.; Zhou, X.; Wang, W.; Qiu, Q.; Huang, X.; Zhou, J.; et al. Convergent Evolution of Rumen Microbiomes in High-Altitude Mammals. Curr. Biol. 2016, 26, 1873–1879. [Google Scholar] [CrossRef]
- Wallace, R.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, 8391–8394. [Google Scholar] [CrossRef] [PubMed]
- Difford, G.F.; Plichta, D.R.; Løvendahl, P.; Lassen, J.; Noel, S.J.; Højberg, O.; Wright, A.D.G.; Zhu, Z.; Kristensen, L.; Nielsen, H.B.; et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLoS Genet. 2018, 14, e1007580. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Gruninger, R.J.; Ribeiro, G.O.; Cameron, A.; McAllister, T.A. Invited review: Application of meta-omics to understand the dynamic nature of the rumen microbiome and how it responds to diet in ruminants. Animal 2019, 13, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- de Assis Lage, C.F.; Räisänen, S.E.; Melgar, A.; Nedelkov, K.; Chen, X.; Oh, J.; Fetter, M.E.; Indugu, N.; Bender, J.S.; Vecchiarelli, B.; et al. Comparison of Two Sampling Techniques for Evaluating Ruminal Fermentation and Microbiota in the Planktonic Phase of Rumen Digesta in Dairy Cows. Front. Microbiol. 2020, 11, 618032. [Google Scholar] [CrossRef] [PubMed]
- Gianesella, M.; Morgante, M.; Stelletta, C.; Ravarotto, L.; Giudice, E.; van Saun, R.J. Evaluating the effects of rumenocentesis on health and performance in dairy cows. Acta. Vet. Brno. 2010, 79, 459–468. [Google Scholar] [CrossRef][Green Version]
- Hagey, J.V.; Laabs, M.; Maga, E.A.; DePeters, E.J. Rumen sampling methods bias bacterial communities observed. PLoS ONE 2022, 17, e0258176. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, M.; Zhang, R.; Zhu, W.; Mao, S. Comparative studies of the composition of bacterial microbiota associated with the ruminal content, ruminal epithelium and in the faeces of lactating dairy cows. Microb. Biotechnol. 2016, 9, 257–268. [Google Scholar] [CrossRef]
- Mohammadzadeh, H.; Yáñez-Ruiz, D.R.; Martínez-Fernandez, G.; Abecia, L. Molecular comparative assessment of the microbial ecosystem in rumen and faeces of goats fed alfalfa hay alone or combined with oats. Anaerobe 2014, 29, 52–58. [Google Scholar] [CrossRef]
- Frey, J.C.; Pell, A.N.; Berthiaume, R.; Lapierre, H.; Lee, S.; Ha, J.K.; Mendell, J.E.; Angert, E.R. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows. J. Appl. Microbiol. 2010, 108, 1982–1993. [Google Scholar] [CrossRef]
- Tapio, I.; Shingfield, K.J.; McKain, N.; Bonin, A.; Fischer, D.; Bayat, A.R.; Vilkki, J.; Taberlet, P.; Snelling, T.J.; Wallace, R.J. Oral samples as non-invasive proxies for assessing the composition of the rumen microbial community. PLoS ONE 2016, 11, e0151220. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.J.; Olijhoek, D.W.; Mclean, F.; Lovendahl, P.; Lund, P.; Hojberg, O. Rumen and Fecal Microbial Community Structure ofHolstein and Jersey Dairy Cows as Affected by Breed, Diet, and Residual Feed Intake. Animals 2019, 9, 498. [Google Scholar] [CrossRef]
- Callaway, T.R.; Dowd, S.E.; Edrington, T.S.; Anderson, R.C.; Krueger, N.; Bauer, N.; Kononoff, P.J.; Nisbet, D.J. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J. Anim. Sci. 2010, 88, 3977–3983. [Google Scholar] [CrossRef] [PubMed]
- Kotz, A.; Azevedo, P.A.; Khafipour, E.; Plaizier, J.C. Effects of the dietary grain content on rumen and fecal microbiota of dairy cows. Can. J. Anim. Sci. 2021, 101, 274–286. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, H.; Wang, Y.; Cao, Z.; Yang, H.; Li, S. Effect of limit-fed diets with different forage to concentrate ratios on fecal bacterial and archaeal community composition in Holstein heifers. Front. Microbiol. 2018, 9, 976. [Google Scholar] [CrossRef] [PubMed]
- de Jesus-Laboy, K.M.; Godoy-Vitorino, F.; Piceno, Y.M.; Tom, L.M.; Pantoja-Feliciano, I.G.; Rivera-Rivera, M.J.; Andersen, G.L.; Domínguez-Bello, M.G. Comparison of the fecal microbiota in feral and domestic goats. Genes 2012, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Lin, X.; Wang, Z.; Hou, Q.; Wang, Y.; Hu, Z. High-production dairy cattle exhibit different rumen and fecal bacterial community and rumen metabolite profile than low-production cattle. Microbiologyopen 2019, 8, e00673. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.L.; Indugu, N.; Vecchiarelli, B.; Bender, J.; Pappalardo, C.; Leibstein, M.; Toth, J.; Katepalli, A.; Garapati, S.; Pitta, D. Temporal changes in the fecal bacterial community in Holstein dairy calves from birth through the transition to a solid diet. PLoS ONE 2020, 15, e0238882. [Google Scholar] [CrossRef]
- Meale, S.J.; Li, S.; Azevedo, P.; Derakhshani, H.; Plaizier, J.C.; Khafipour, E.; Steele, M.A. Development of ruminal and fecal microbiomes are affected by weaning but not weaning strategy in dairy calves. Front. Microbiol. 2016, 7, 582. [Google Scholar] [CrossRef]
- Hodgson, J.; Forbes, T.D.A.; Armstrong, R.H.; Beattie, M.M.; Hunter, E.A. Comparative Studies of the Ingestive Behaviour and Herbage Intake of Sheep and Cattle Grazing Indigenous Hill Plant. Communities. J. Appl. Ecol. 1991, 28, 205–227. [Google Scholar] [CrossRef]
- Clark, D.A.; Lambert, M.G.; Rolston, M.P.; Dymock, N. Diet selection by goats and sheep on hill country. Proc. New Zeal. Soc. Anim. Prod. 1985, 42, 155–157. [Google Scholar]
- Ming, L.; Yi, L.; Siriguleng; Hasi, S.; He, J.; Hai, L.; Wang, Z.; Guo, F.; Qiao, X. Jirimutu Comparative analysis of fecal microbial communities in cattle and Bactrian camels. PLoS ONE 2017, 12, e0173062. [Google Scholar] [CrossRef] [PubMed]
- Mott, A.C.; Schneider, D.; Hünerberg, M.; Hummel, J.; Tetens, J. Bovine Rumen Microbiome: Impact of DNA Extraction Methods and Comparison of Non-Invasive Sampling Sites. Ruminants 2022, 2, 112–132. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Hagey, J.V.; Bhatnagar, S.; Heguy, J.M.; Karle, B.M.; Price, P.L.; Meyer, D.; Maga, E.A. Fecal microbial communities in a large representative cohort of California dairy cows. Front. Microbiol. 2019, 10, 1093. [Google Scholar] [CrossRef] [PubMed]
- Shanks, O.C.; Kelty, C.A.; Archibeque, S.; Jenkins, M.; Newton, R.J.; McLellan, S.L.; Huse, S.M.; Sogin, M.L. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl. Environ. Microbiol. 2011, 77, 2992–3001. [Google Scholar] [CrossRef]
- Mao, S.; Zhang, R.; Wang, D.; Zhu, W. The diversity of the fecal bacterial community and its relationship with the concentration of volatile fatty acids in the feces during subacute rumen acidosis in dairy cows. BMC Vet. Res. 2012, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ji, S.; Wang, F.; Huang, J.; Alugongo, G.M.; Li, S. Dynamic changes of the fecal bacterial community in dairy cows during early lactation. AMB Express 2020, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.G.N.; Bressani, F.A.; Cuadrat, R.R.C.; Tizioto, P.C.; De Oliveira, P.S.N.; Mourão, G.B.; Coutinho, L.L.; Reecy, J.M.; Koltes, J.E.; Walsh, P.; et al. The structure of microbial populations in Nelore GIT reveals inter-dependency of methanogens in feces and rumen. J. Anim. Sci. Biotechnol. 2020, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Boukerb, A.M.; Noël, C.; Quenot, E.; Cadiou, B.; Chevé, J.; Quintric, L.; Cormier, A.; Dantan, L.; Gourmelon, M. Comparative Analysis of Fecal Microbiomes From Wild Waterbirds to Poultry, Cattle, Pigs, and Wastewater Treatment Plants for a Microbial Source Tracking Approach. Front. Microbiol. 2021, 12, 697553. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, P.; Zhang, K.; Zhang, L.; Wang, X.; Li, L.; Zhang, H. Distinct Stage Changes in Early-Life Colonization and Acquisition of the Gut Microbiota and Its Correlations With Volatile Fatty Acids in Goat Kids. Front. Microbiol. 2020, 11, 584742. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Zhang, T.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; et al. Comparative analysis of the gut microbiota composition between captive and wild forest musk deer. Front. Microbiol. 2017, 8, 1705. [Google Scholar] [CrossRef]
- La Reau, A.J.; Suen, G. The Ruminococci: Key symbionts of the gut ecosystem. J. Microbiol. 2018, 56, 199–208. [Google Scholar] [CrossRef]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and Comparison of Microbiota in the Gastrointestinal Tracts of the Goat (Capra hircus) During Preweaning Development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef]
- Gilbert, M.S.; Pantophlet, A.J.; Berends, H.; Pluschke, A.M.; Van den Borne, J.J.G.C.; Hendriks, W.H.; Schols, H.A.; Gerrits, W.J.J. Fermentation in the small intestine contributes substantially to intestinal starch disappearance in calves. J. Nutr. 2015, 145, 1147–1155. [Google Scholar] [CrossRef]
- Liu, J.; Bian, G.; Sun, D.; Zhu, W.; Mao, S. Starter feeding supplementation alters colonic mucosal bacterial communities and modulates mucosal immune homeostasis in newborn lambs. Front. Microbiol. 2017, 8, 429. [Google Scholar] [CrossRef]
- Abu Aboud, O.A.; Adaska, J.M.; Williams, D.R.; Rossitto, P.V.; Champagne, J.D.; Lehenbauer, T.W.; Atwill, R.; Li, X.; Aly, S.S. Epidemiology of Salmonella sp. in California cull dairy cattle: Prevalence of fecal shedding and diagnostic accuracy of pooled enriched broth culture of fecal samples. PeerJ 2016, 4, e2386. [Google Scholar] [CrossRef] [PubMed]
- Shabana, I.I.; Albakri, N.N.; Bouqellah, N.A. Metagenomic investigation of faecal microbiota in sheep and goats of the same ages. J. Taibah Univ. Sci. 2021, 15, 1–9. [Google Scholar] [CrossRef]
- Khiaosa-ard, R.; Zebeli, Q. Diet-induced inflammation: From gut to metabolic organs and the consequences for the health and longevity of ruminants. Res. Vet. Sci. 2018, 120, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ye, H.; Liu, J.; Mao, S. High-grain diets altered rumen fermentation and epithelial bacterial community and resulted in rumen epithelial injuries of goats. Appl. Microbiol. Biotechnol. 2017, 101, 6981–6992. [Google Scholar] [CrossRef] [PubMed]
- Plaizier, J.C.; Danscher, A.M.; Azevedo, P.A.; Derakhshani, H.; Andersen, P.H.; Khafipour, E. A grain-based sara challenge affects the composition of epimural and mucosa-associated bacterial communities throughout the digestive tract of dairy cows. Animals 2021, 11, 1658. [Google Scholar] [CrossRef]
- Mu, Y.; Qi, W.; Zhang, T.; Zhang, J.; Mao, S. Multi-omics Analysis Revealed Coordinated Responses of Rumen Microbiome and Epithelium to High-Grain-Induced Subacute Rumen Acidosis in Lactating Dairy Cows. Msystems 2022, 7, e01490-21. [Google Scholar] [CrossRef]
- Hume, I.D. Concepts of Digestive Efficiency. In Physiological Ecology; Starck, J.M., Wang, T., Eds.; Science Publishers: Enfield, NH, USA, 2005; pp. 43–58. ISBN 1-57808-246-3. [Google Scholar]
- Silanikove, N. The physiological basis of adaptation in goats to harsh environments. Small Rumin. Res. 2000, 35, 181–193. [Google Scholar] [CrossRef]
- Giger-Reverdin, S.; Domange, C.; Broudiscou, L.P.; Sauvant, D.; Berthelot, V. Rumen function in goats, an example of adaptive capacity. J. Dairy Res. 2020, 87, 45–51. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PERMANOVA p-Value (* p < 0.05) | PERMDISP p-Value (* p < 0.05) | |
|---|---|---|
| Bray curtis distance | 0.001 ** | 0.01 * |
| Jaccard distance | 0.001 ** | 0.002 * |
| Weighted unifrac distance | 0.001 ** | 0.02 * |
| Unweighted unifrac distance | 0.046 * | 0.04 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahayri, T.M.; Fliegerová, K.O.; Mattiello, S.; Celozzi, S.; Mrázek, J.; Mekadim, C.; Sechovcová, H.; Kvasnová, S.; Atallah, E.; Moniello, G. Host Species Affects Bacterial Evenness, but Not Diversity: Comparison of Fecal Bacteria of Cows and Goats Offered the Same Diet. Animals 2022, 12, 2011. https://doi.org/10.3390/ani12162011
Mahayri TM, Fliegerová KO, Mattiello S, Celozzi S, Mrázek J, Mekadim C, Sechovcová H, Kvasnová S, Atallah E, Moniello G. Host Species Affects Bacterial Evenness, but Not Diversity: Comparison of Fecal Bacteria of Cows and Goats Offered the Same Diet. Animals. 2022; 12(16):2011. https://doi.org/10.3390/ani12162011
Chicago/Turabian StyleMahayri, Tiziana Maria, Kateřina Olša Fliegerová, Silvana Mattiello, Stefania Celozzi, Jakub Mrázek, Chahrazed Mekadim, Hana Sechovcová, Simona Kvasnová, Elie Atallah, and Giuseppe Moniello. 2022. "Host Species Affects Bacterial Evenness, but Not Diversity: Comparison of Fecal Bacteria of Cows and Goats Offered the Same Diet" Animals 12, no. 16: 2011. https://doi.org/10.3390/ani12162011
APA StyleMahayri, T. M., Fliegerová, K. O., Mattiello, S., Celozzi, S., Mrázek, J., Mekadim, C., Sechovcová, H., Kvasnová, S., Atallah, E., & Moniello, G. (2022). Host Species Affects Bacterial Evenness, but Not Diversity: Comparison of Fecal Bacteria of Cows and Goats Offered the Same Diet. Animals, 12(16), 2011. https://doi.org/10.3390/ani12162011