
Whole-Genome Survey Analyses Provide a New Perspective for the Evolutionary Biology of Shimofuri Goby, Tridentiger bifasciatus

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Genome Survey Sequencing
2.2. Data Analysis Methods
3. Results
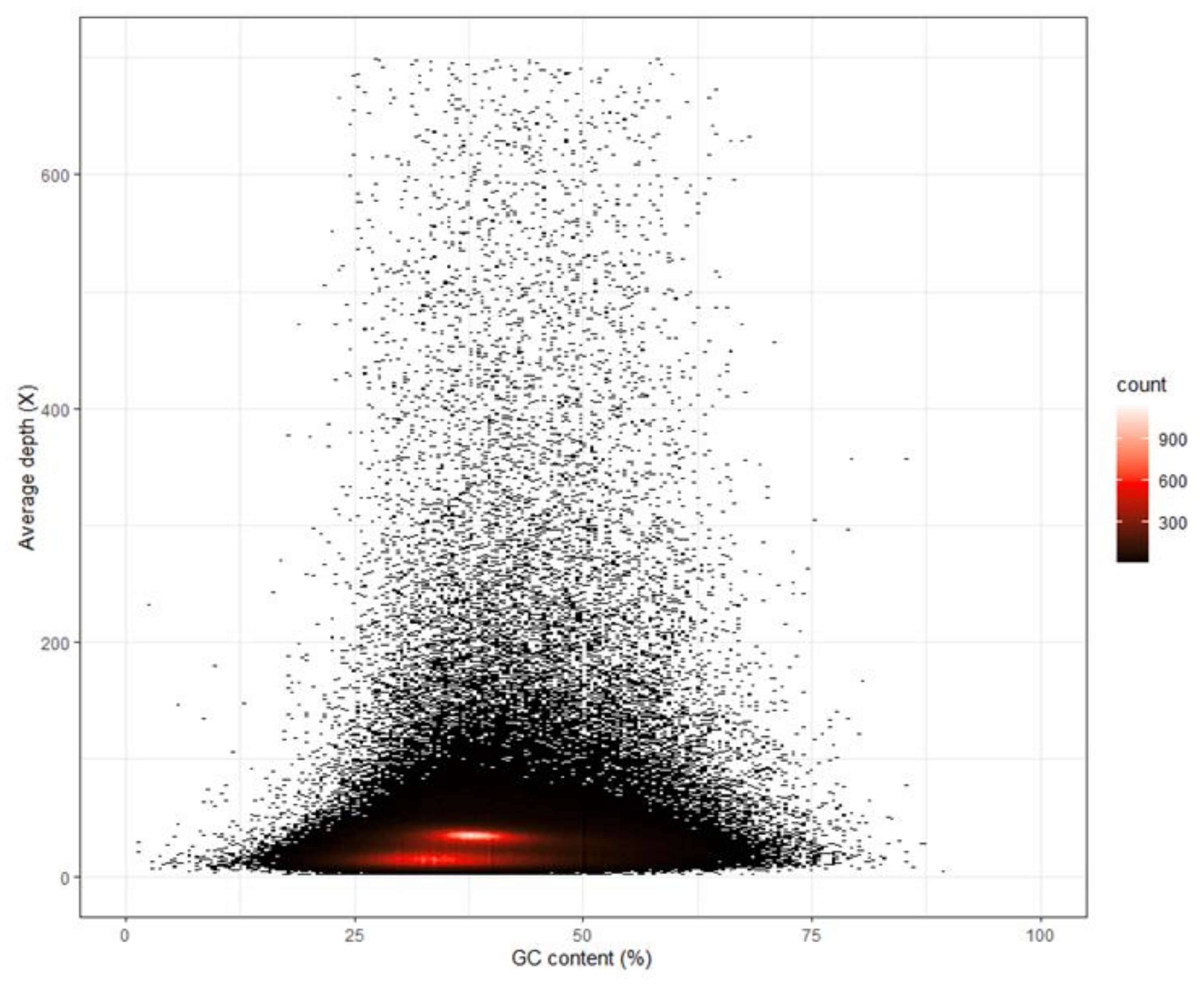
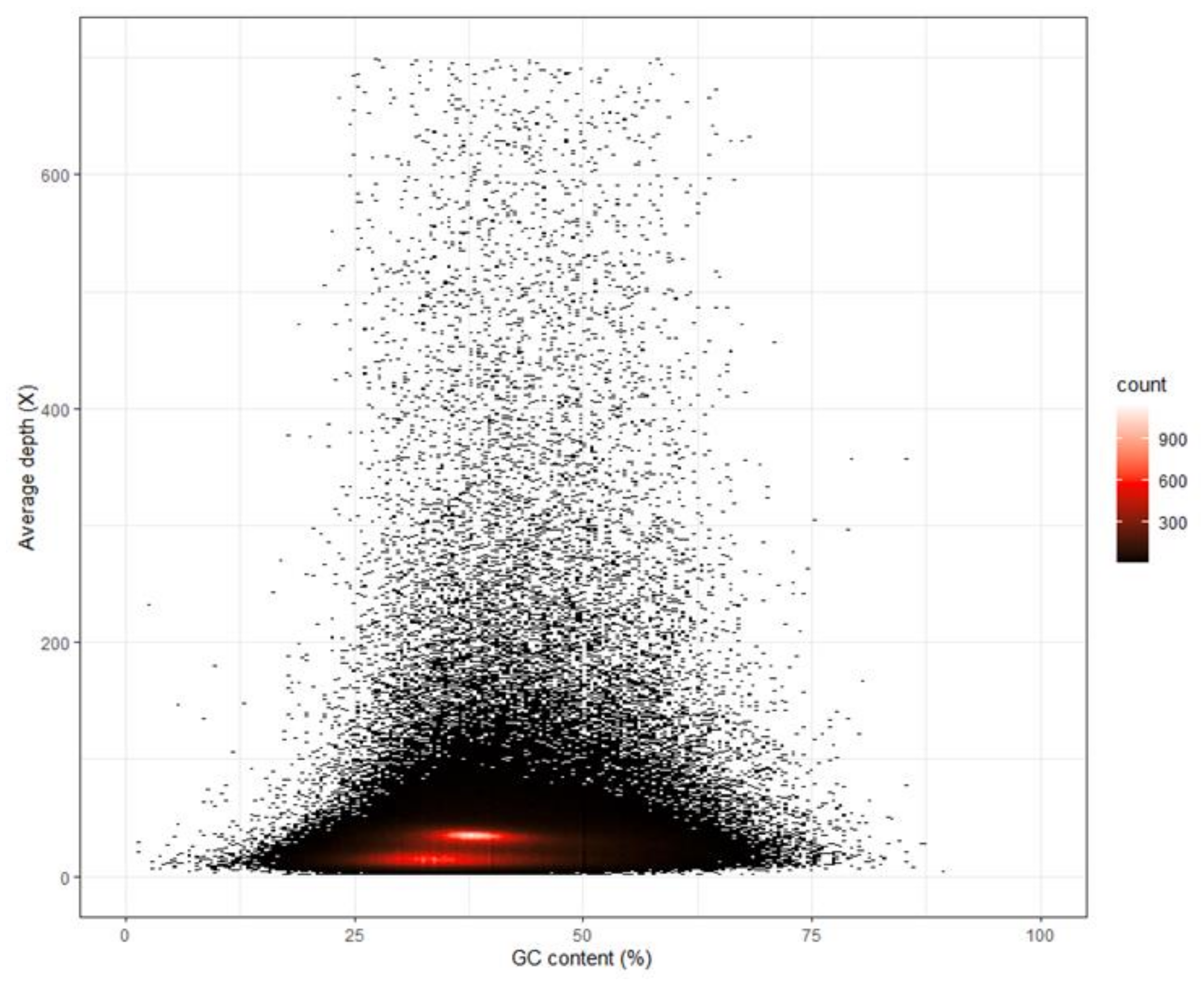
3.1. Whole-Genome Sequencing, K-Mer Analysis and Genome Assembly
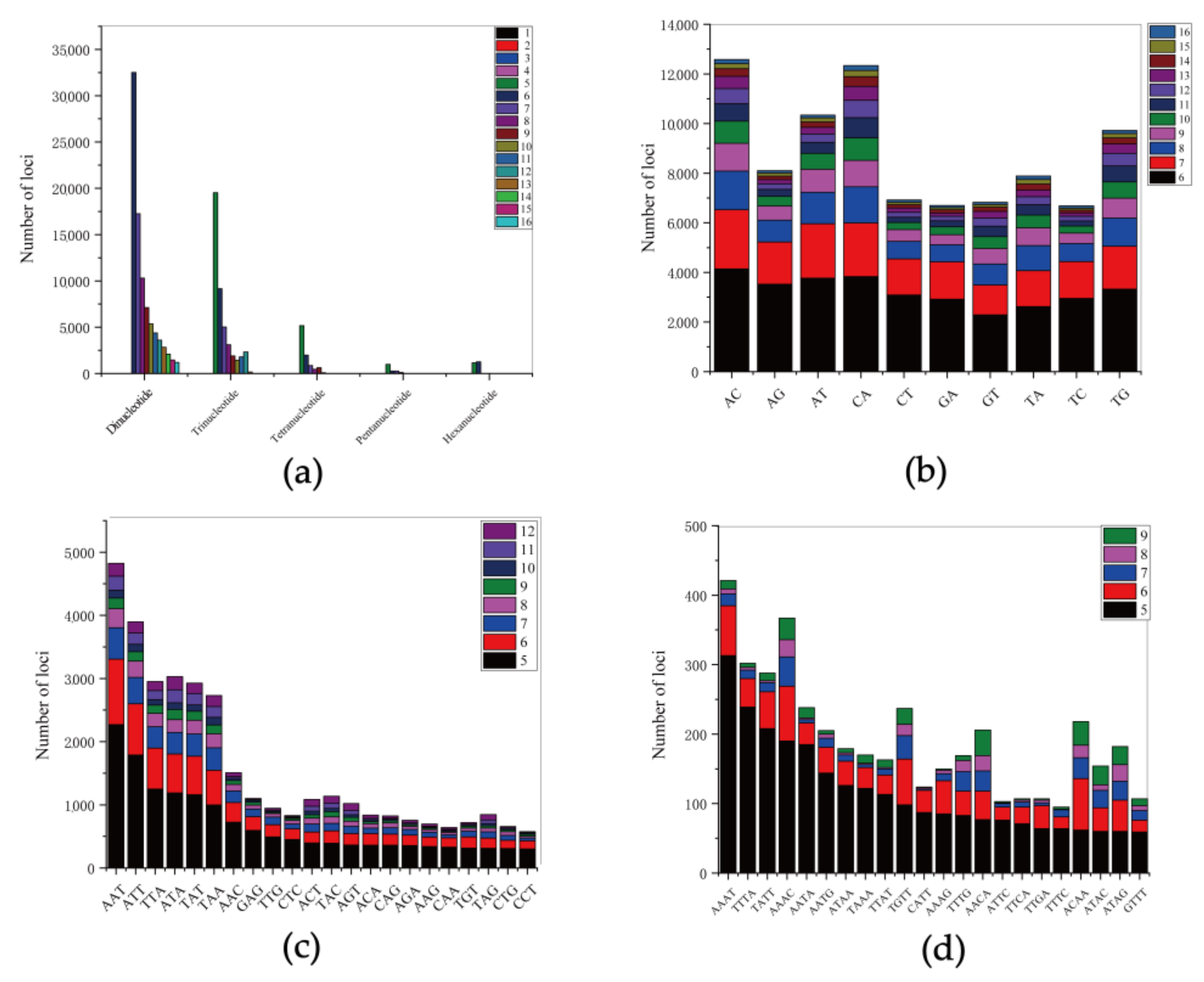
3.2. Identification and Characterization of SSR for the Genome of the Shimofuri Goby
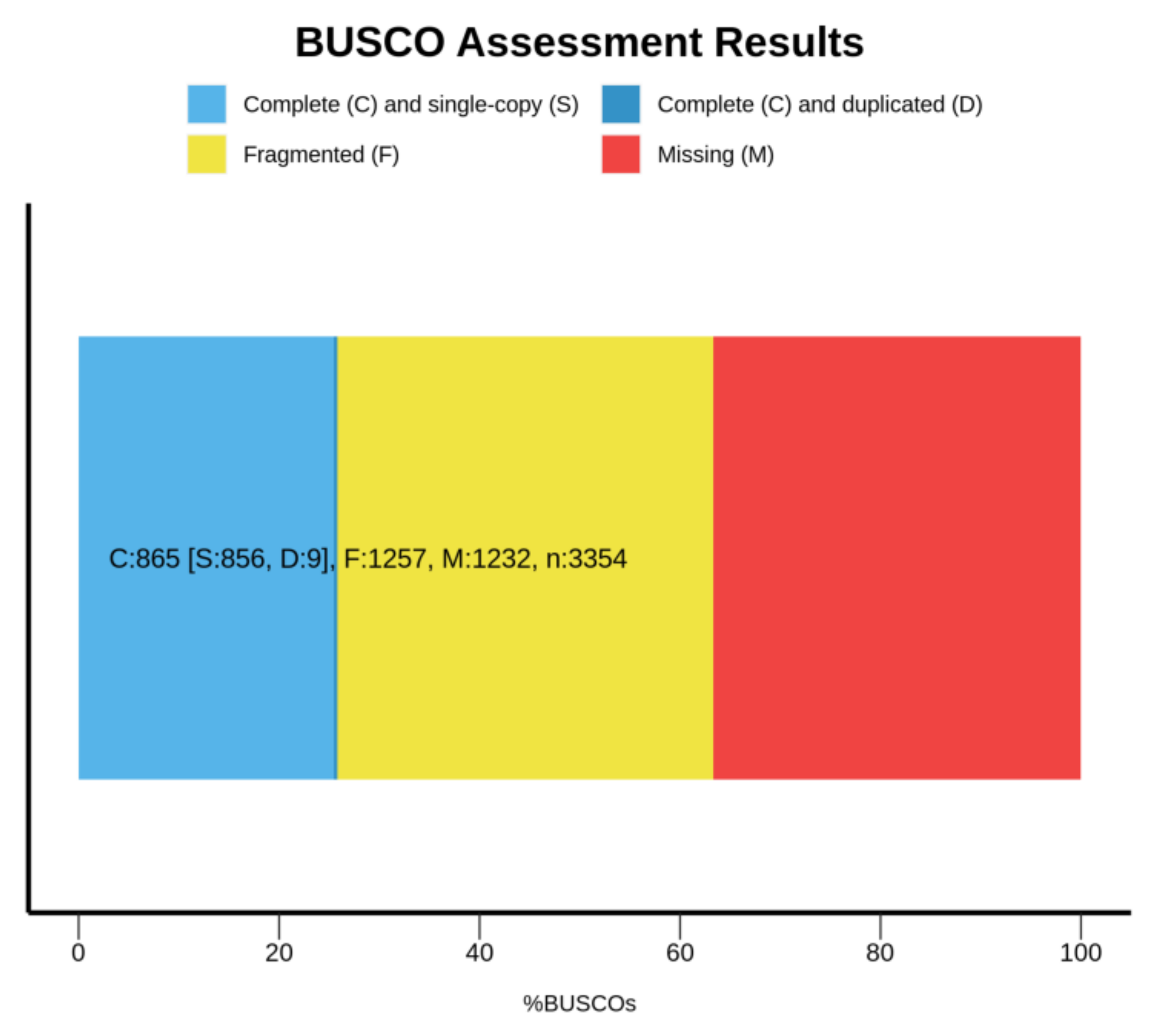
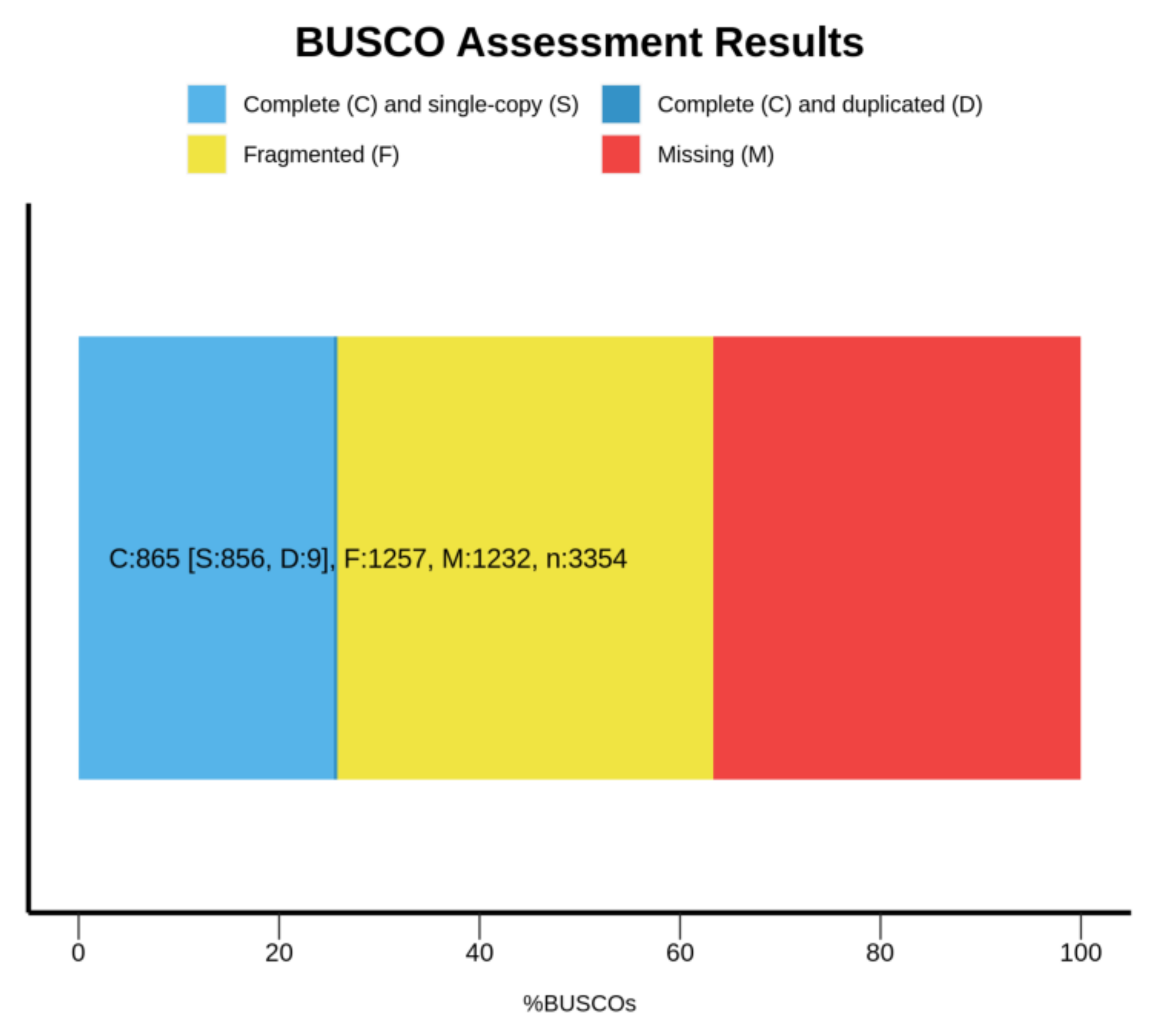
3.3. Identification of Single-Copy Homologous Genes in the Shimofuri Goby Genome
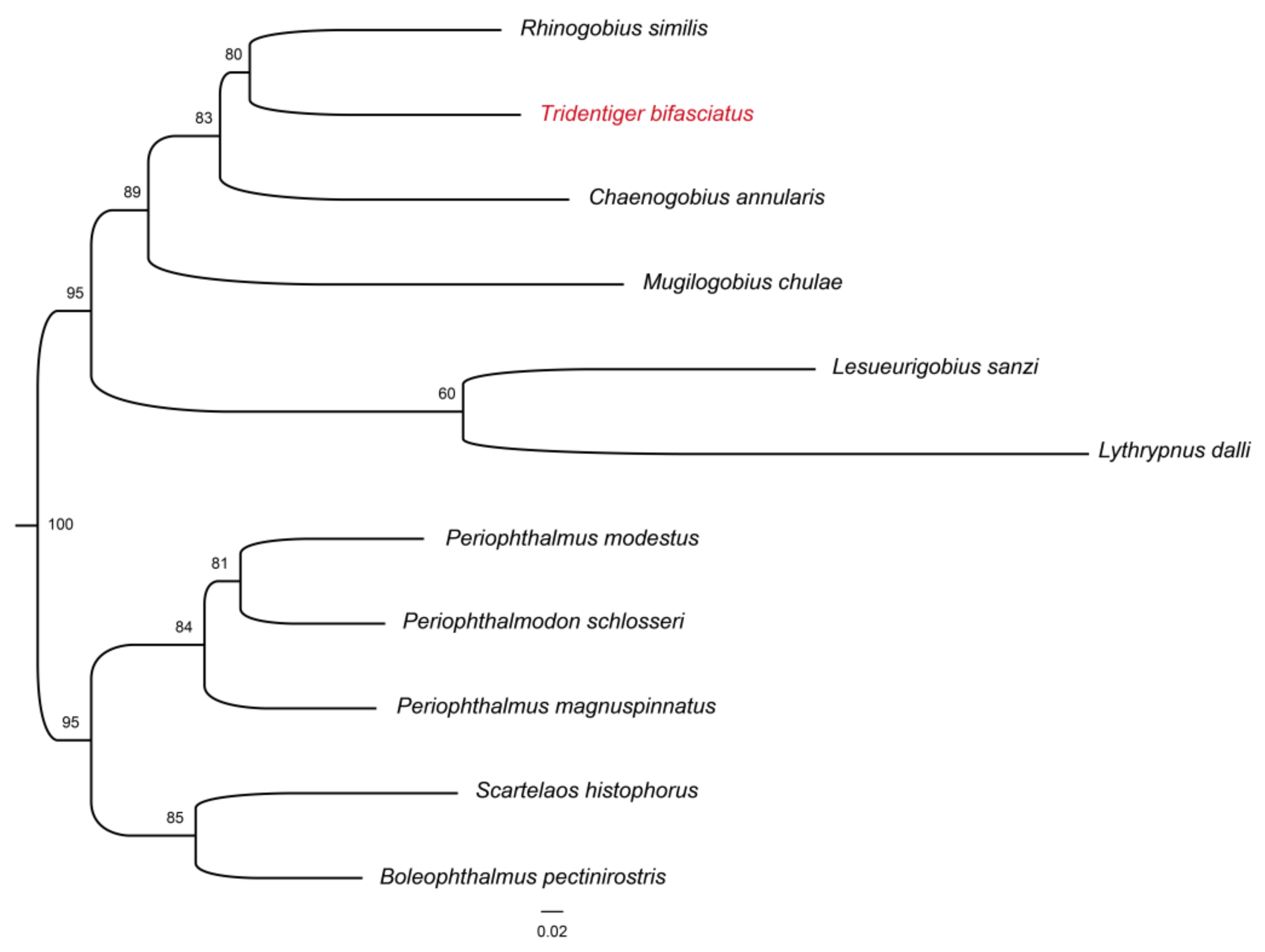
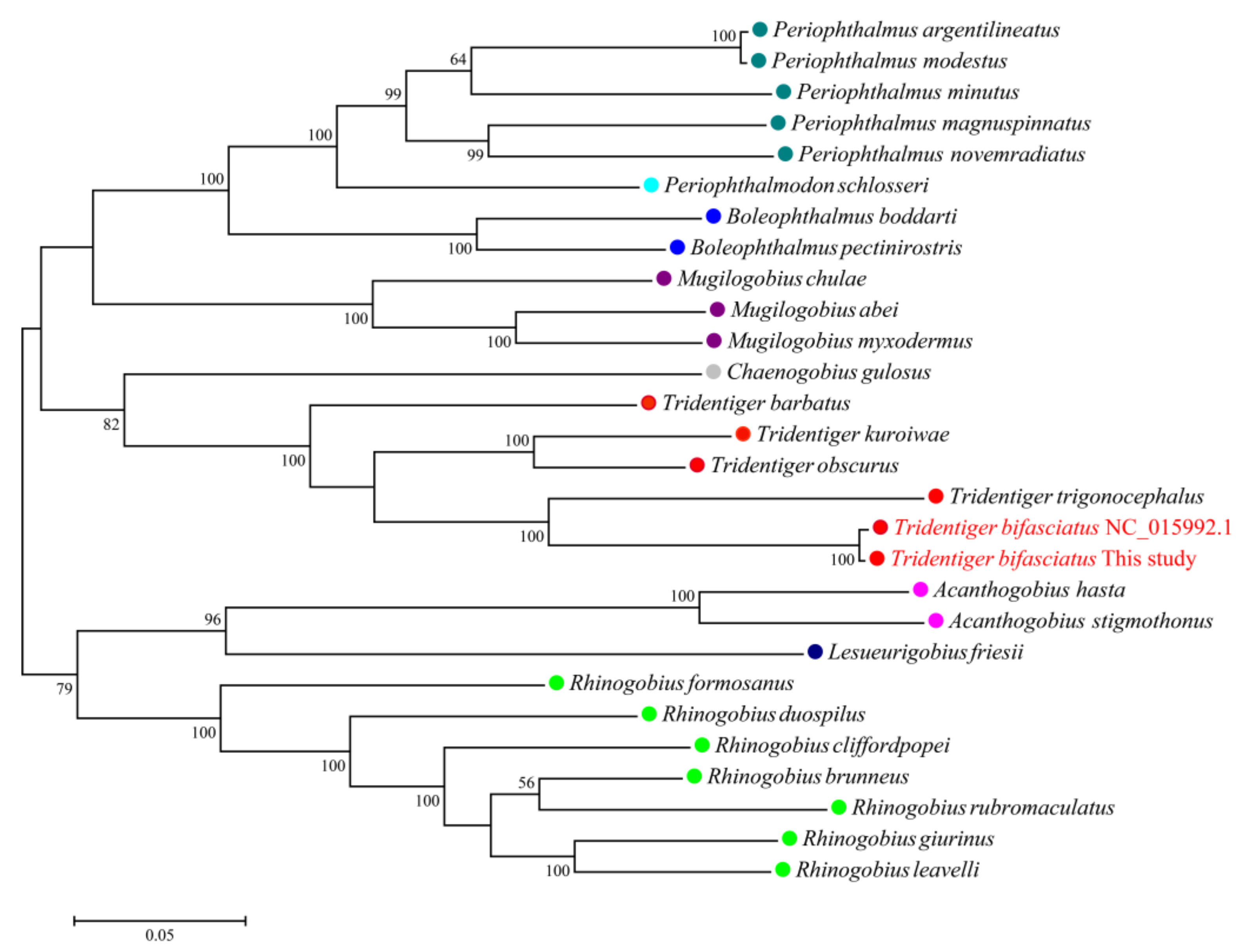
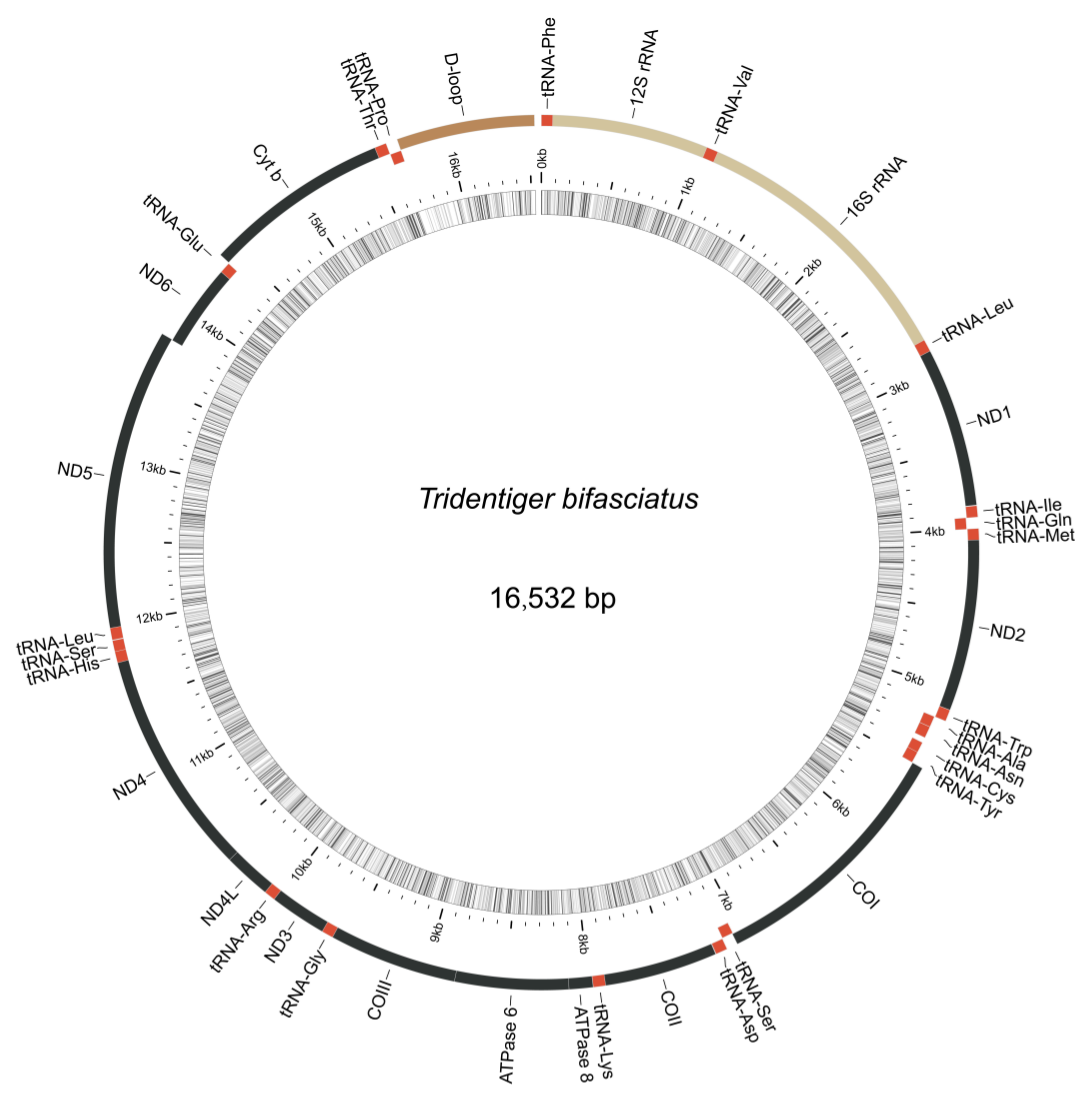
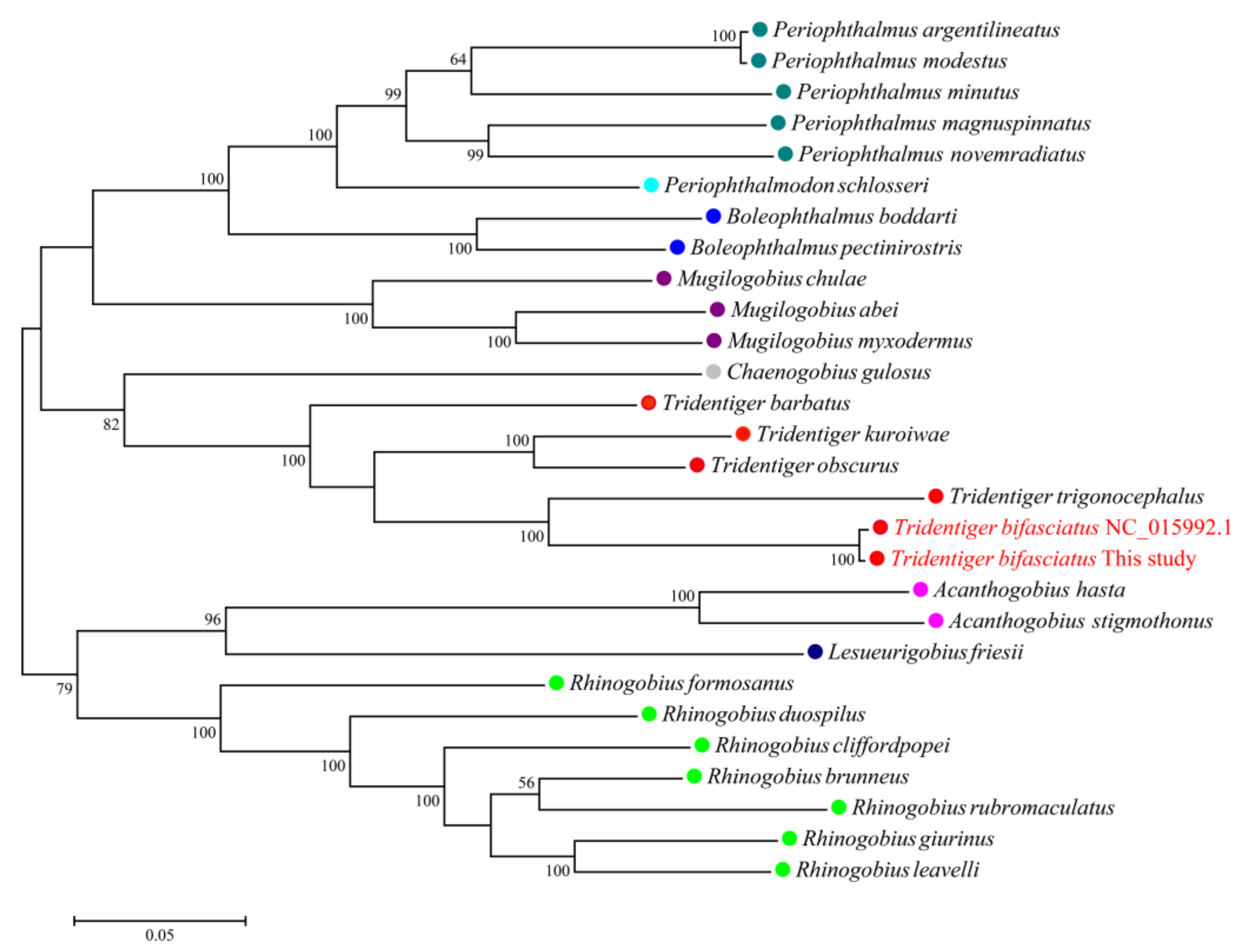
3.4. Mitochondrial Genome Assembly of the Shimofuri Goby and Phylogenetic Analysis
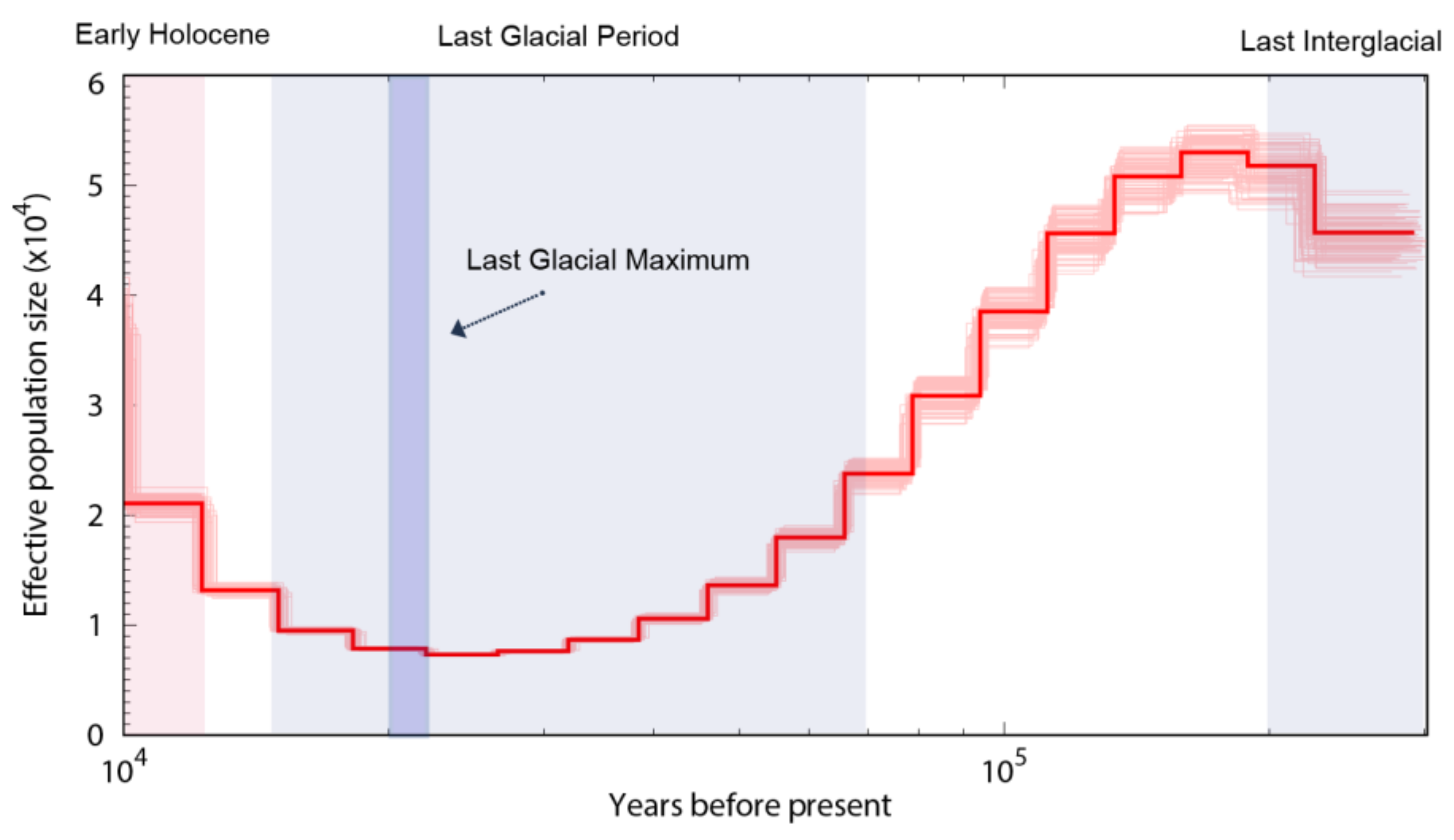
3.5. Population Size Dynamics of the Shimofuri Goby
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meng, L.; Moyle, P.B.; Herbold, B. Changes in abundance and distribution of native and introduced fishes of suisun marsh. Trans. Am. Fish. Soc. 1994, 123, 498–507. [Google Scholar] [CrossRef]
- Sakamoto, K. Reexamination of the status of the striped goby. Jpn. J. Ichthyol. 1989, 36, 100–112. [Google Scholar] [CrossRef]
- O’grady, K.; Cowx, I. Nonindigenous fishes introduced into inland waters of the united states. Fish. Manag. Ecol. 2000, 2, 172–175. [Google Scholar] [CrossRef]
- Matern, S.A.; Moyle, P.B.; Pierce, L.C. Native and alien fishes in a california estuarine marsh: Twenty-one years of changing assemblages. Trans. Am. Fish. Soc. 2002, 131, 797–816. [Google Scholar] [CrossRef]
- Matern, S.A.; Fleming, K.J. Invasion of a third Asian goby, Tridentiger bifasciatus, into California. Calif. Fish. Game 1995, 81, 71–76. [Google Scholar]
- Matern, S.A. Using temperature and salinity tolerances to predict the success of the shimofuri goby, a recent invader into california. Trans. Am. Fish. Soc. 2001, 130, 592–599. [Google Scholar] [CrossRef]
- Qin, J.; Xiang, M.; Jia, M.; Cheng, F.; Xie, S. Combined opportunistic and equilibrium life-history traits facilitate successful invasions of the shimofuri goby (Tridentiger bifasciatus). Aquat. Invasions 2020, 15. in press. [Google Scholar]
- Kim, H.; Yu, N.L.; Jeong, J.M.; Kim, H.J.; Baeck, G.W. Diet composition of juvenile Trachurus japonicus in the coastal waters of geumodo yeosu, korea. Bull. Korean Soc. Fish. Technol. 2015, 51, 637–643. [Google Scholar] [CrossRef]
- Qin, J.; Xie, S.; Cheng, F. Broad diet composition and seasonal feeding variation facilitate successful invasion of the shimofuri goby (Tridentiger bifasciatus) in a water transfer system. Water 2020, 12, 3411. [Google Scholar] [CrossRef]
- Jin, X.; Wang, R.; Wei, T.; Tang, D.; Xu, T. Complete mitochondrial genome sequence of Tridentiger bifasciatus and Tridentiger barbatus (Perciformes, Gobiidae): A mitogenomic perspective on the phylogenetic relationships of Gobiidae. Mol. Biol. Rep. 2015, 42, 253–265. [Google Scholar] [CrossRef]
- Raphalo, E.M.; Gu, C.; Lü, Z.; Chen, Y. Genetic structure of intertidal shimofuri goby in the zhejiang coastal sea, china: Implications for management. Mar. Biodivers. 2018, 49, 1–9. [Google Scholar] [CrossRef]
- Qin, J.; Victor, S.B.; Cheng, F.; Xie, S. Development and characterization of 14 novel microsatellite markers for an invasive goby (Tridentiger bifasciatus) in water transfer system. J. Appl. Ichthyol. 2021, 37, 314–317. [Google Scholar]
- Chen, B.; Sun, Z.; Lou, F.; Gao, T.X.; Song, N. Genomic characteristics and profile of microsatellite primers for Acanthogobius ommaturus by genome survey sequencing. Biosci. Rep. 2020, 40, BSR20201295. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, H.; Gao, T. Comprehensive whole genome survey analyses of male and female brown-spotted flathead fish Platycephalus sp.1. Genomics 2020, 112, 4742–4748. [Google Scholar] [CrossRef]
- Bi, Q.; Zhao, Y.; Cui, Y.; Wang, L. Genome survey sequencing and genetic background characterization of yellow horn based on next-generation sequencing. Mol. Biol. Rep. 2019, 46, 4303–4312. [Google Scholar] [CrossRef]
- Lu, M.; An, H.; Li, L. Genome Survey Sequencing for the Characterization of the Genetic Background of Rosa roxburghii Tratt and Leaf Ascorbate Metabolism Genes. PLoS ONE 2016, 11, e0147530. [Google Scholar] [CrossRef] [PubMed]
- Ziya Motalebipour, E.; Kafkas, S.; Khodaeiaminjan, M.; Çoban, N.; Gözel, H. Genome survey of pistachio (Pistacia vera L.) by next generation sequencing: Development of novel SSR markers and genetic diversity in Pistacia species. BMC Genom. 2016, 17, 998. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Zhang, Y.X.; Yang, M.J.; Sun, J.C.; Zhang, T.; Wang, H.Y. Genome survey on invasive veined rapa whelk (Rapana venosa) and development of microsatellite loci on large scale. J. Genet. 2018, 97, e79–e86. [Google Scholar] [CrossRef]
- Gilbert, P.S.; Chang, J.; Pan, C.; Sobel, E.M.; Sinsheimer, J.S.; Faircloth, B.C.; Alfaro, M.E. Genome-wide ultraconserved elements exhibit higher phylogenetic informativeness than traditional gene markers in percomorph fishes. Mol. Phylogenetics Evol. 2015, 92, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, M.H.; Henderson, J.B.; Esposito, L.; Trautwein, M. Genomic Characterization and Curation of UCEs Improves Species Tree Reconstruction. Syst. Biol. 2021, 70, 307–321. [Google Scholar] [CrossRef]
- Koprek, T.; Rangel, S.; McElroy, D.; Louwerse, J.D.; Williams-Carrier, R.E.; Lemaux, P.G. Transposon-mediated single-copy gene delivery leads to increased transgene expression stability in barley. Plant Physiol. 2001, 125, 1354–1362. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quant. Biol. 2013, 35, 62–67. [Google Scholar]
- Allio, R.; Schomaker-Bastos, A.; Romiguier, J.; Prosdocimi, F.; Nabholz, B.; Delsuc, F. MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 2020, 20, 892–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Liu, G.; Wei, Y.; Zhu, Y.; Li, J.; Miao, Z.; Chen, M.; Yue, Z.; Yu, L.; Dong, Z.; et al. Whole-genome sequencing reveals sex determination and liver high-fat storage mechanisms of yellow stripe goby (Mugilogobius chulae). Commun. Biol. 2021, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Bian, C.; Zan, Q.; Xu, X.; Liu, X.; Chen, J.; Wang, J.; Qiu, Y.; Li, W.; Zhang, X.; et al. Mudskipper genomes provide insights into the terrestrial adaptation of amphibious fishes. Nat. Commun. 2014, 5, 5594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, F.; Han, M.; Peng, Z. Evolution and diversity of transposable elements in fish genomes. Sci. Rep. 2019, 9, 15399. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Xu, S.; Han, Z.; Liu, Q.; Ke, W.; Liu, A.; Gao, T. Chromosome-Level Genome Assembly and Annotation of a Sciaenid Fish, Argyrosomus japonicus. Genome Biol. Evol. 2021, 13, evaa246. [Google Scholar] [CrossRef]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef]
- Jo, E.; Lee, S.J.; Choi, E.; Kim, J.; Lee, S.G.; Lee, J.H.; Kim, J.H.; Park, H. Whole genome survey and microsatellite motif identification of Artemia franciscana. Biosci. Rep. 2021, 41, BSR20203868. [Google Scholar] [CrossRef]
- Jo, E.; Cho, Y.H.; Lee, S.J.; Choi, E.; Kim, J.; Kim, J.H.; Chi, Y.M.; Park, H. Genome survey and microsatellite motif identification of Pogonophryne albipinna. Biosci. Rep. 2021, 41, BSR20210824. [Google Scholar] [CrossRef]
- Harr, B.; Schl ötterer, C. Long microsatellite alleles in Drosophila melanogaster have a downward mutation bias and short persistence times, which cause their genome-wide underrepresentation. Genetics 2000, 155, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, D.; Li, M.; Mustapha, U.F.; Tian, C.; Chen, H.; Huang, Y.; Deng, S.; Wu, T.; Zhu, C.; et al. Genome Survey of Male and Female Spotted Scat (Scatophagus argus). Animals 2019, 9, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Feulner, P.; Eizaguirre, C.; Lenz, T.L.; Bornberg-Bauer, E.; Milinski, M.; Reusch, T.; Chain, F. Genome-Wide Genotype-Expression Relationships Reveal Both Copy Number and Single Nucleotide Differentiation Contribute to Differential Gene Expression between Stickleback Ecotypes. Genome Biol. Evol. 2019, 11, 2344–2359. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M. Distinguishing homologous from analogous proteins. Syst. Zool 1970, 19, 99–113. [Google Scholar] [CrossRef]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef]
- Consuegra, S.; John, E.; Verspoor, E.; Leaniz, C.D. Patterns of natural selection acting on the mitochondrial genome of a locally adapted fish species. Genet. Sel. Evol. 2015, 47, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, A.; Glasgow, J.; Jaschke, P.R.; Bawazer, L.A.; Munson, M.S.; Cochran, J.R.; Endy, D.; Salit, M. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 2017, 45, 3615–3626. [Google Scholar] [CrossRef] [Green Version]
- Morel, B.; Schade, P.; Lutteropp, S.; Williams, T.A.; Szöllősi, G.J.; Stamatakis, A. SpeciesRax: A Tool for Maximum Likelihood Species Tree Inference from Gene Family Trees under Duplication, Transfer, and Loss. Mol. Biol. Evol. 2022, 39, msab365. [Google Scholar] [CrossRef]
- Martin, A.P.; Kessing, B.D.; Palumbi, S.R. Accuracy of estimating genetic distance between species from short sequences of mitochondrial DNA. Mol. Biol. Evol. 1990, 7, 485–488. [Google Scholar]
- Canales-Aguirre, C.B.; Ferrada-Fuentes, S.; Galleguillos, R.; Oyarzun, F.X.; Buratti, C.C.; Hernández, C.E. High genetic diversity and low-population differentiation in the Patagonian sprat (Sprattus fuegensis) based on mitochondrial DNA. Mitochondrial DNA. Part. A 2018, 29, 1148–1155. [Google Scholar] [CrossRef]
- Debes, P.V.; Zachos, F.E.; Hanel, R. Mitochondrial phylogeography of the European sprat (Sprattus sprattus L., Clupeidae) reveals isolated climatically vulnerable populations in the Mediterranean Sea and range expansion in the northeast Atlantic. Mol. Ecol. 2008, 17, 3873–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latta, L.C.; Fisk, D.L.; Knapp, R.A.; Pfrender, M.E. Genetic resilience of daphnia populations following experimental removal of introduced fish. Conserv. Genet. 2010, 11, 1737–1745. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Length (bp) | Total Number | Max Length (bp) | N50 Length (bp) | N90 Length (bp) | |
|---|---|---|---|---|---|
| Contig | 830,520,379 | 2,382,088 | 25,967 | 691 | 127 |
| Scaffold | 818,382,753 | 1,814,709 | 42,478 | 1203 | 127 |
| Statistical Items | Numbers |
|---|---|
| Total number of sequences examined | 1,814,709 |
| Total number of identified SSRs | 288,730 |
| Number of sequences containing more than 1 SSR | 48,813 |
| Number of SSRs present in compound formation | 30,529 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Liu, Y.; Du, X.; Ma, S.; Song, N.; Zhao, L. Whole-Genome Survey Analyses Provide a New Perspective for the Evolutionary Biology of Shimofuri Goby, Tridentiger bifasciatus. Animals 2022, 12, 1914. https://doi.org/10.3390/ani12151914
Zhao X, Liu Y, Du X, Ma S, Song N, Zhao L. Whole-Genome Survey Analyses Provide a New Perspective for the Evolutionary Biology of Shimofuri Goby, Tridentiger bifasciatus. Animals. 2022; 12(15):1914. https://doi.org/10.3390/ani12151914
Chicago/Turabian StyleZhao, Xiang, Yaxian Liu, Xueqing Du, Siyu Ma, Na Song, and Linlin Zhao. 2022. "Whole-Genome Survey Analyses Provide a New Perspective for the Evolutionary Biology of Shimofuri Goby, Tridentiger bifasciatus" Animals 12, no. 15: 1914. https://doi.org/10.3390/ani12151914
APA StyleZhao, X., Liu, Y., Du, X., Ma, S., Song, N., & Zhao, L. (2022). Whole-Genome Survey Analyses Provide a New Perspective for the Evolutionary Biology of Shimofuri Goby, Tridentiger bifasciatus. Animals, 12(15), 1914. https://doi.org/10.3390/ani12151914