New Evidence of Tiger Subspecies Differentiation and Environmental Adaptation: Comparison of the Whole Genomes of the Amur Tiger and the South China Tiger


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Construction of a Genome Library
2.3. Resequencing and Variant Identification
2.4. Copy Number Variation
2.5. Calculation of LD
2.6. Selective Sweep Analysis
3. Results
3.1. Sequencing and Variation
3.2. The Divergence between the Amur Tiger and the South China Tiger
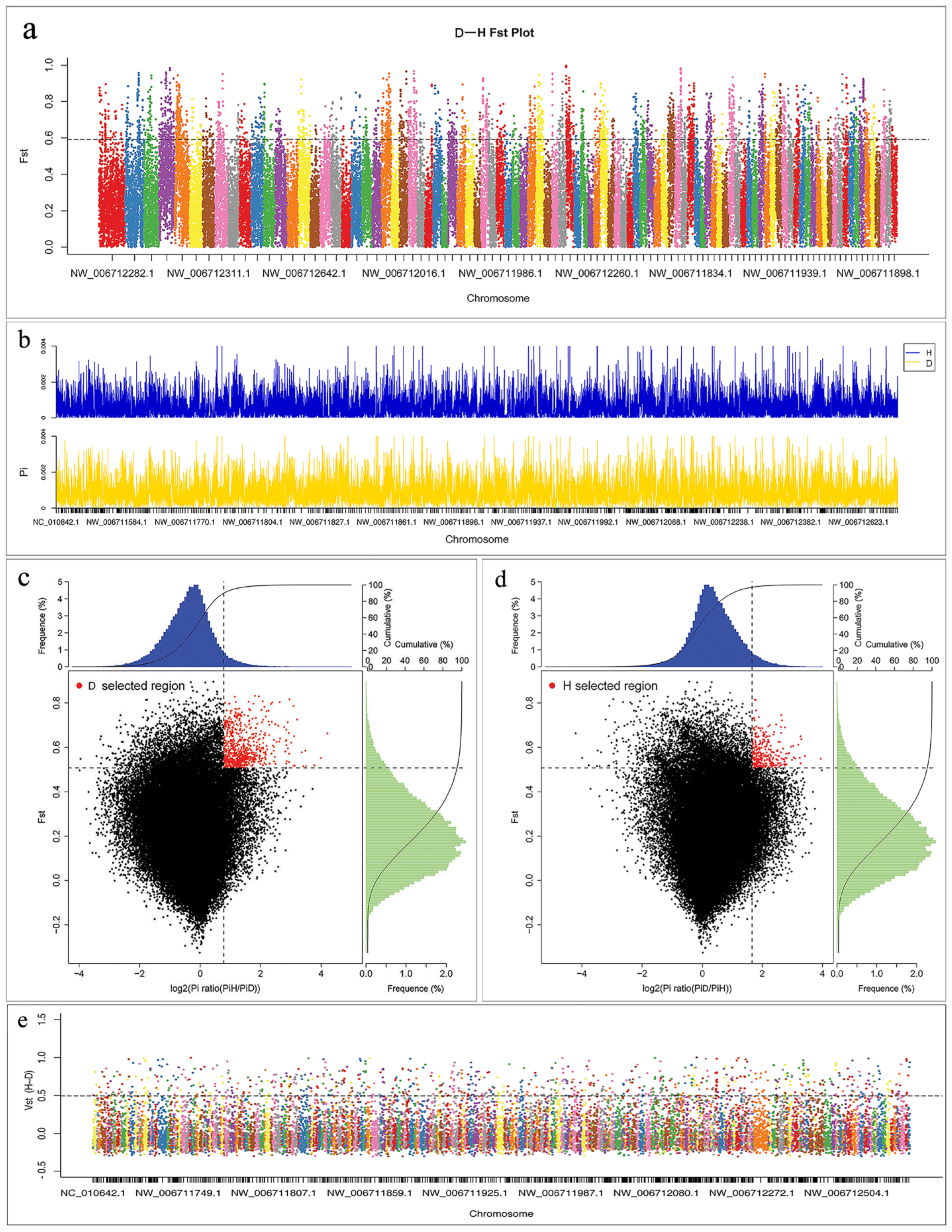
3.3. Screening and Annotation of Selective Sweeps based on SNP
3.4. Selective Elimination Analysis based on CNV
3.5. The Differentiation Mechanism of Tiger Subspecies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luo, S.-J.; Kim, J.-H.; Johnson, W.E.; van der Walt, J.; Martenson, J.; Yuhki, N.; Miquelle, D.G.; Uphyrkina, O.; Goodrich, J.M.; Quigley, H.B. Phylogeography and Genetic Ancestry of Tigers (Panthera tigris). PLoS Biol. 2004, 2, e442. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.; Lynam, A.; Miquelle, D.; Wibisono, H.; Kawanishi, K.; Pattanavibool, A.; Htun, S.; Tempa, T.; Karki, J.; Jhala, Y.; et al. Panthera tigris. In IUCN Red List of Threatened Species; IUCN: Gland, Switzerland, 2015; p. e.T15955A50659951. [Google Scholar]
- Smith, J.L.D.; Ahearn, S.C.; McDougal, C. Landscape Analysis of Tiger Distribution and Habitat Quality in Nepal. Conserv. Biol. 1998, 12, 1338–1346. [Google Scholar] [CrossRef]
- Driscoll, C.A.; Yamaguchi, N.; Bar-Gal, G.K.; Roca, A.L.; Luo, S.; Macdonald, D.W.; O’Brien, S.J. Mitochondrial Phylogeography Illuminates the Origin of the Extinct Caspian Tiger and Its Relationship to the Amur Tiger. PLoS ONE 2009, 4, e4125. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.-J.; Johnson, W.E.; O’Brien, S.J. Applying Molecular Genetic Tools to Tiger Conservation. Integr. Zool. 2010, 5, 351–362. [Google Scholar] [CrossRef]
- King, T. Saving the South China Tiger. Scientist 2007, 21, 16. [Google Scholar]
- Yin, Y.Z. Studbook of the South China Tiger; Chongqing Zoological Garden: Chongqing, China, 2004. [Google Scholar]
- Xu, Y.C.; Fang, S.G.; Li, Z.K. Sustainability of the South China Tiger: Implications of Inbreeding Depression and Introgression. Conserv. Genet. 2007, 8, 1199–1207. [Google Scholar] [CrossRef]
- O’Brien, S.J. A Role for Molecular Genetics in Biological Conservation. Proc. Natl. Acad. Sci. USA 1994, 91, 5748–5755. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, H.N.; Hayssen, V.; Bruford, M.W. Conservation of Deer: Contributions from Molecular Biology, Evolutionary Ecology, and Reproductive Physiology. J. Zool. 1997, 243, 461–484. [Google Scholar] [CrossRef]
- Procaccini, G.; Olsen, J.L.; Reusch, T.B. Contribution of Genetics and Genomics to Seagrass Biology and Conservation. J. Exp. Mar. Biol. Ecol. 2007, 350, 234–259. [Google Scholar] [CrossRef]
- Komoroske, L.M.; Jensen, M.P.; Stewart, K.R.; Shamblin, B.M.; Dutton, P.H. Advances in the Application of Genetics in Marine Turtle Biology and Conservation. Front. Mar. Sci. 2017, 4, 156. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Yin, Y.; Liu, Q. Inbreeding Depression and Population Viability Analysis of the South China Tigers (Panthera tigris amoyensis) in Captivity. Mamm. Biol. 2021, 101, 803–809. [Google Scholar] [CrossRef]
- Traylor-Holzer, K.; Zhong, X.; Yuzhong, Y. The Struggle to Save the Last South China Tigers. In Tigers of The World; William Andrew Publishing: Amsterdam, The Netherlands, 2010; pp. 457–461. [Google Scholar]
- Zhang, L.; Lan, T.; Lin, C.; Fu, W.; Yuan, Y.; Lin, K.; Li, H.; Kumar Sahu, S.; Liu, Z.; Chen, D. Chromosome-scale Genomes Reveal Genomic Consequences of Inbreeding in the South China Tiger: A Comparative Study with the Amur Tiger. Mol. Ecol. Resour. 2022. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Funk, W.C.; Rajora, O.P. Population Genomics for Wildlife Conservation and Management. Mol. Ecol. 2021, 30, 62–82. [Google Scholar] [CrossRef]
- Qin, Y.; Nyhus, P.J.; Larson, C.L.; Carroll, C.J.; Muntifering, J.; Dahmer, T.D.; Jun, L.; Tilson, R.L. An Assessment of South China Tiger Reintroduction Potential in Hupingshan and Houhe National Nature Reserves, China. Biol. Conserv. 2015, 182, 72–86. [Google Scholar] [CrossRef] [Green Version]
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the Future of Conservation Genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Johnson, W.E.; Onorato, D.P.; Roelke, M.E.; Land, E.D.; Cunningham, M.; Belden, R.C.; McBride, R.; Jansen, D.; Lotz, M.; Shindle, D. Genetic Restoration of the Florida Panther. Science 2010, 329, 1641–1645. [Google Scholar] [CrossRef]
- Cho, Y.S.; Hu, L.; Hou, H.; Lee, H.; Xu, J.; Kwon, S.; Oh, S.; Kim, H.-M.; Jho, S.; Kim, S. The Tiger Genome and Comparative Analysis with Lion and Snow Leopard Genomes. Nat. Commun. 2013, 4, 2433. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, K. Genomic Variant Annotation and Prioritization with ANNOVAR and WANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural Variation in the Human Genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An Approach to Discover, Genotype, and Characterize Typical and Atypical CNVs from Family and Population Genome Sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubitzky, W.; Wolkenhauer, O.; Cho, K.-H.; Yokota, H. Encyclopedia of Systems Biology; Springer: New York, NY, USA, 2013. [Google Scholar]
- Jae-Heup, K.; Eizirik, E.; O’Brien, S.J.; Johnson, W.E. Structure and Patterns of Sequence Variation in the Mitochondrial DNA Control Region of the Great Cats. Mitochondrion 2001, 1, 279–292. [Google Scholar] [CrossRef]
- Zhang, W.P.; Zhang, Z.H.; Shen, F.J.; Yue, B.S. Mitochondrial D-Loop and NADH Dehydrogenase Subunit 5 (NDS) Gene of Panthera tigris amoyensis, P. tigris altaica and P. tigris tigris and Their Application in Phylogenetic Analysis. Sichuan J. Zool. 2005, 24, 22. [Google Scholar]
- Shi, J.; Tian, C.; Xu, J.; Zhang, X.; Li, L.; Zhang, S. Applications of Mitochondrial Genes to an Analysis of the Phylogenetic Relationship among Tigers. J. S. China Agric. Univ. 2014, 35, 13–46. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 1, 1358–1370. [Google Scholar]
- Mockenhaupt, F.P.; Cramer, J.P.; Hamann, L.; Stegemann, M.S.; Eckert, J.; Oh, N.-R.; Otchwemah, R.N.; Dietz, E.; Ehrhardt, S.; Schröder, N.W. Toll-like Receptor (TLR) Polymorphisms in African Children: Common TLR-4 Variants Predispose to Severe Malaria. Proc. Natl. Acad. Sci. USA 2006, 103, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Pedurupillay, C.R.J.; Landsend, E.C.S.; Vigeland, M.D.; Ansar, M.; Frengen, E.; Misceo, D.; Strømme, P. Segregation of Incomplete Achromatopsia and Alopecia Due to PDE6H and LPAR6 Variants in a Consanguineous Family from Pakistan. Genes 2016, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Hassall, M.M.; Barnard, A.R.; MacLaren, R.E. Focus: Genome Editing: Gene Therapy for Color Blindness. Yale J. Biol. Med. 2017, 90, 543. [Google Scholar] [PubMed]
- Zillikens, M.C.; Demissie, S.; Hsu, Y.-H.; Yerges-Armstrong, L.M.; Chou, W.-C.; Stolk, L.; Livshits, G.; Broer, L.; Johnson, T.; Koller, D.L. Large Meta-Analysis of Genome-Wide Association Studies Identifies Five Loci for Lean Body Mass. Nat. Commun. 2017, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Patton-Vogt, J. Transport and Metabolism of Glycerophosphodiesters Produced through Phospholipid Deacylation. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2007, 1771, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Blaya, C.; Moorjani, P.; Salum, G.A.; Gonçalves, L.; Weiss, L.A.; Leistner-Segal, S.; Manfro, G.G.; Smoller, J.W. Preliminary Evidence of Association between EFHC2, a Gene Implicated in Fear Recognition, and Harm Avoidance. Neurosci. Lett. 2009, 452, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Contreras, A.; Postlethwait, J.H. Evolution of the MiR199-214 Cluster and Vertebrate Skeletal Development. RNA Biol. 2014, 11, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Kitamoto, A.; Kitamoto, T.; Nakamura, T.; Matsuo, T.; Nakata, Y.; Hyogo, H.; Ochi, H.; Kamohara, S.; Miyatake, N.; Kotani, K. CDH13 Polymorphisms Are Associated with Adiponectin Levels and Metabolic Syndrome Traits Independently of Visceral Fat Mass. J. Atheroscler. Thromb. 2015, 23, 31567. [Google Scholar] [CrossRef] [Green Version]
- Chikamoto, K.; Misu, H.; Takayama, H.; Kikuchi, A.; Ishii, K.; Lan, F.; Takata, N.; Tajima-Shirasaki, N.; Takeshita, Y.; Tsugane, H. Rapid Response of the Steatosis-Sensing Hepatokine LECT2 during Diet-Induced Weight Cycling in Mice. Biochem. Biophys. Res. Commun. 2016, 478, 1310–1316. [Google Scholar] [CrossRef]
- Göddeke, S.; Knebel, B.; Fahlbusch, P.; Hörbelt, T.; Poschmann, G.; Van de Velde, F.; Benninghoff, T.; Al-Hasani, H.; Jacob, S.; Van Nieuwenhove, Y. CDH13 Abundance Interferes with Adipocyte Differentiation and Is a Novel Biomarker for Adipose Tissue Health. Int. J. Obes. 2018, 42, 1039–1050. [Google Scholar] [CrossRef]
- Zhao, J.; Mommersteeg, M.T. Slit–Robo Signalling in Heart Development. Cardiovasc. Res. 2018, 114, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.T. The Application of Ecological Rules to the Racial Anthropology of the Aboriginal New World. Am. Anthropol. 1953, 55, 311–327. [Google Scholar] [CrossRef]
- Benian, G.M.; Kiff, J.E.; Neckelmann, N.; Moerman, D.G.; Waterston, R.H. Sequence of an Unusually Large Protein Implicated in Regulation of Myosin Activity in C. Elegans. Nature 1989, 342, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.P.; Somlyo, A.V. Signal Transduction and Regulation in Smooth Muscle. Nature 1994, 372, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in Mice Lacking the Gene for Endothelial Nitric Oxide Synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.-S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated Blood Pressures in Mice Lacking Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, R.; Gappa, N.; Rutz, M.; Schlossmann, J.; Rose, C.R.; Konnerth, A.; Brummer, S.; Kühbandner, S.; Hofmann, F. Functional Reconstitution of Vascular Smooth Muscle Cells with CGMP-Dependent Protein Kinase I Isoforms. Circ. Res. 2002, 90, 1080–1086. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.J.; Brugger, H.; Boyd, J.; Paal, P. Accidental Hypothermia. N. Engl. J. Med. 2012, 367, 1930–1938. [Google Scholar] [CrossRef]
- El Ouaamari, A.; InSug, O.; Shirakawa, J.; Basile, G.; Zhang, W.; Roger, S.; Thomou, T.; Xu, S.; Qiang, G.; Liew, C.W. Forkhead Box Protein O1 (FoxO1) Regulates Hepatic Serine Protease Inhibitor B1 (SerpinB1) Expression in a Non-Cell-Autonomous Fashion. J. Biol. Chem. 2019, 294, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Kousteni, S. FoxO1, the Transcriptional Chief of Staff of Energy Metabolism. Bone 2012, 50, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.T. Biological Adaptation of Man to His Environment: Heat, Cold, Altitude, and Nutrition. Ann. N. Y. Acad. Sci. 1961, 91, 617–633. [Google Scholar] [CrossRef]
- Potts, R. Environmental Hypotheses of Hominin Evolution. Am. J. Phys. Anthropol. 1998, 107, 93–136. [Google Scholar] [CrossRef]
- Hemmer, H. The Phylogeny of the Tiger (Panthera tigris). In Tigers of the World: The Biology, Biopolitics, Management and Conservation of an Endangered Species; Tilson, R.L., Seal, U.S., Eds.; Noyes Publications: Park Ridge, NJ, USA, 1987; pp. 28–35. [Google Scholar]
- Hemmer, H. Wohin Gehört “Felis” Palaeosinensis Zdansky, 1924, in Systematischer Hinsicht. Neues Jahrb. Geol. Paläontol. Abh. 1967, 129, 83–96. [Google Scholar]
- Collier, G.E.; O’Brien, S.J. A Molecular Phylogeny of the Felidae: Immunological Distance. Evolution 1985, 39, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Mazak, V. Panthera tigris. Mamm. Species 1981, 152, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, E.E.; Khan, A.; Taylor, R.W.; Gouy, A.; Greenbaum, G.; Thiéry, A.; Kang, J.T.; Redondo, S.A.; Prost, S.; Barsh, G. Recent Evolutionary History of Tigers Highlights Contrasting Roles of Genetic Drift and Selection. Mol. Biol. Evol. 2021, 38, 2366–2379. [Google Scholar] [CrossRef]
- Liu, S.; Lorenzen, E.D.; Fumagalli, M.; Li, B.; Harris, K.; Xiong, Z.; Zhou, L.; Korneliussen, T.S.; Somel, M.; Babbitt, C. Population Genomics Reveal Recent Speciation and Rapid Evolutionary Adaptation in Polar Bears. Cell 2014, 157, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, X.; Yue, B.; Hou, R.; Xie, J.; Zou, Z.-T.; Han, Y.; Shen, F.; Zhang, L.; Xie, Z. Sorting out the Genetic Background of the Last Surviving South China Tigers. J. Hered. 2019, 110, 641–650. [Google Scholar] [CrossRef]
- Liu, D.; Ma, Y.; Li, H.; Xu, Y.; Zhang, Y.; Dahmer, T.; Bai, S.; Wang, J. Simultaneous Polyandry and Heteropaternity in Tiger (Panthera tigris altaica): Implications for Conservation of Genetic Diversity in Captive Populations of Felids. Chin. Sci. Bull. 2013, 58, 2230–2236. [Google Scholar] [CrossRef] [Green Version]
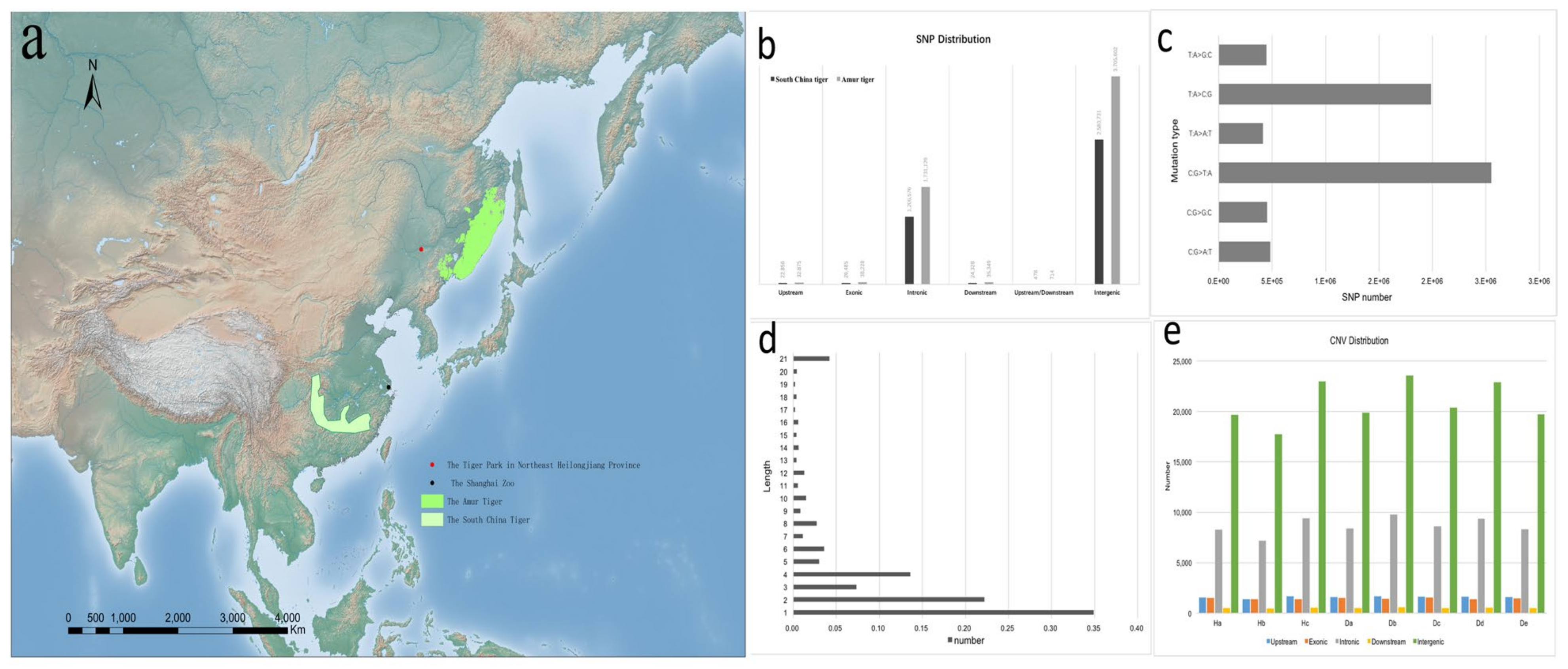
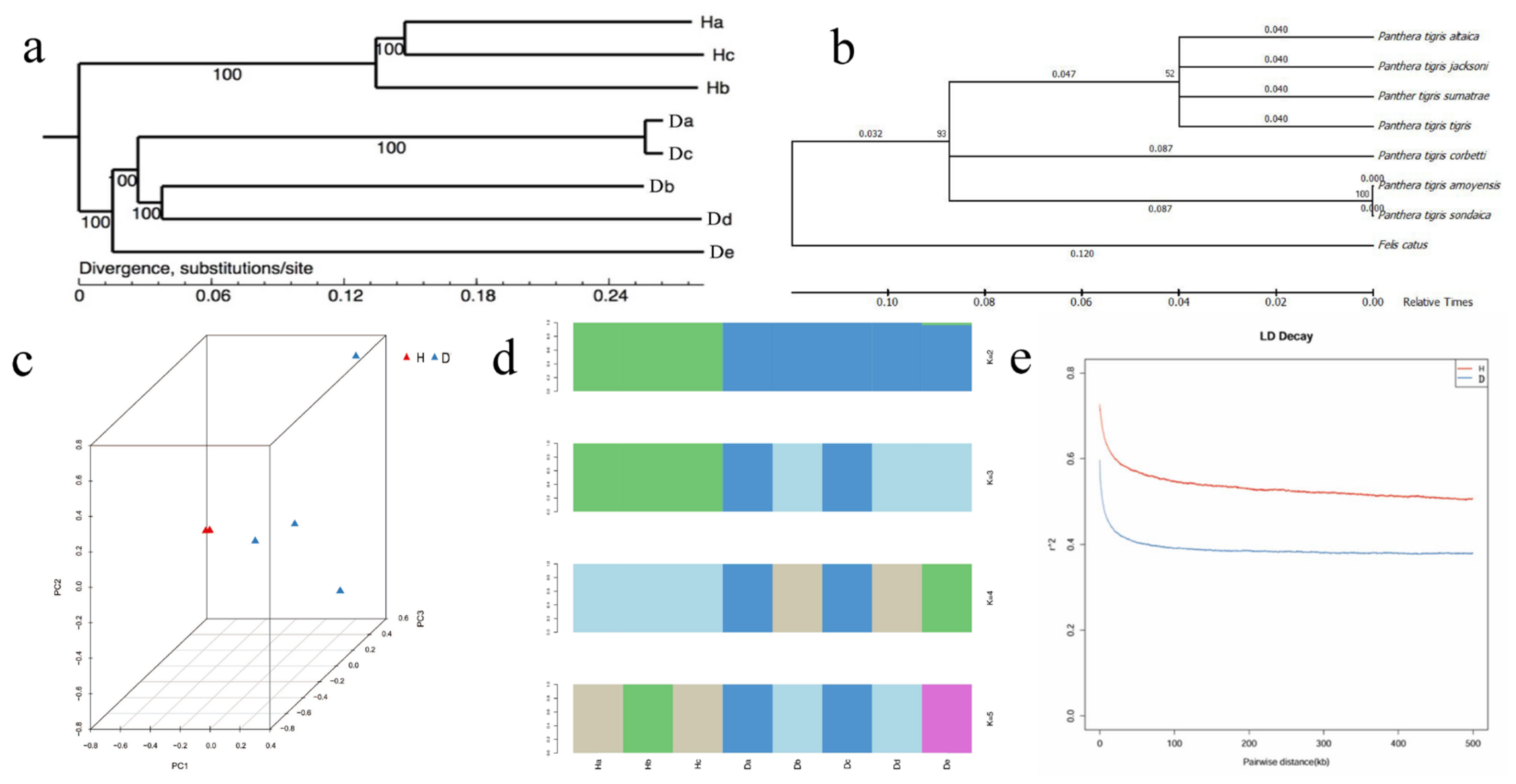

{kind=link}
{kind=link}
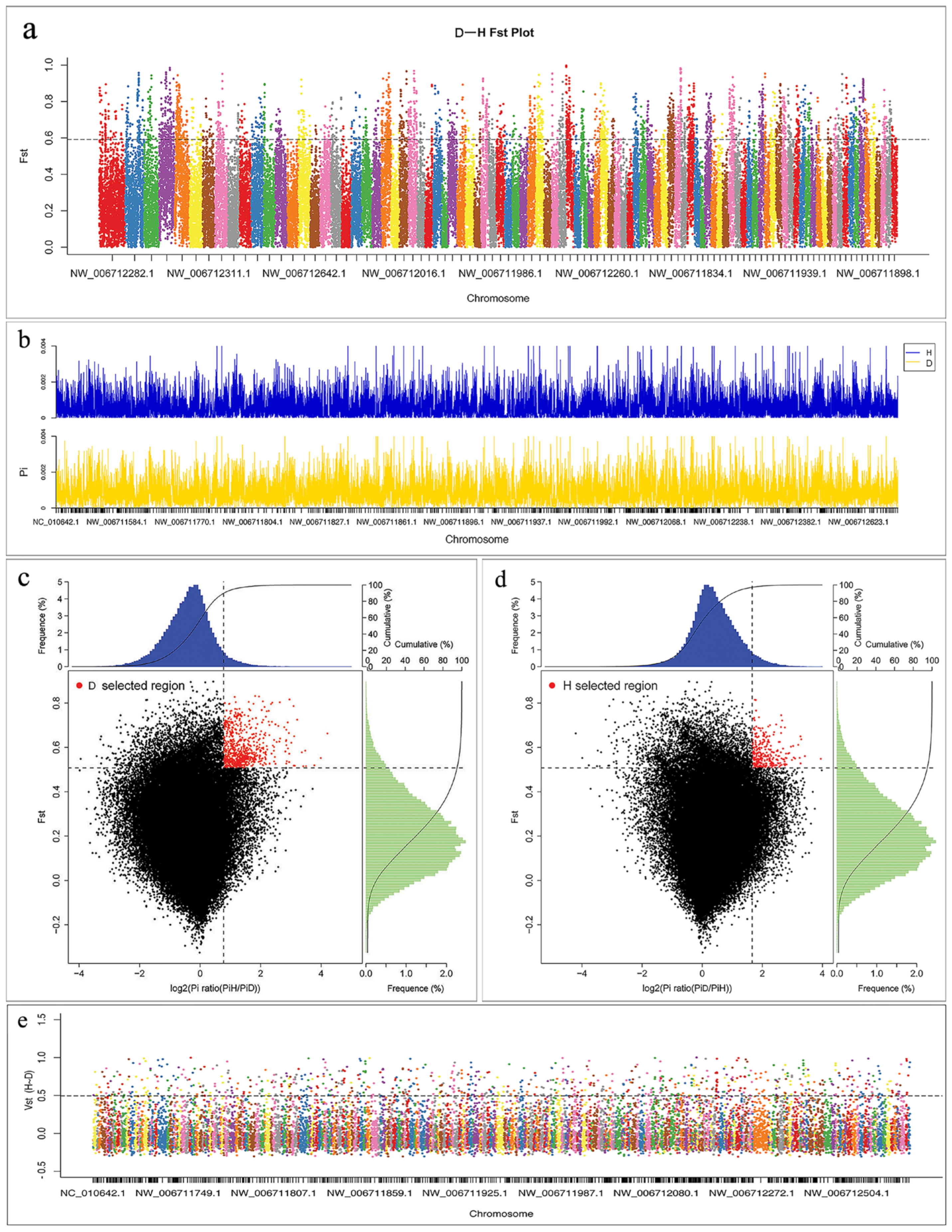{kind=link}
| Genome-Wide | SNP no. | θπ (10–3) | θω (10–3) | Tajima’s D | Non-Syn SNPs | Syn SNPs | Non-Syn/Syn |
|---|---|---|---|---|---|---|---|
| Panthera tigris altaica | 5,579,192 | 0.876 | 0.812 | 0.423 | 16,099 | 21,948 | 0.734 |
| Panthera tigris amoyensis | 3,885,781 | 0.563 | 0.526 | 0.336 | 11,106 | 15,268 | 0.727 |
| Species | Term | Database | ID | Gene Amount | p-Value |
|---|---|---|---|---|---|
| Amur tiger | Ribosome | KEGG PATHWAY | fca03010 | 18 | 0.0287 |
| KEGG ID | Description | Gene Name |
|---|---|---|
| fca05134 | Legionellosis | TLR4, CASP3, UBE2E2, LOC102967742, TRRAP, CNTN1 |
| fca05145 | Toxoplasmosis | LY96, TLR4, CASP3, SFXN1, MAPK8, MAP3K7, IFNGR1, ITGB1 |
| fca05133 | Pertussis | LY96, TLR4, CASP3, MAPK8, LOC102967742, ITGB1 |
| fca05140 | Leishmaniasis | SFXN1, MAP3K7, IFNGR1, TLR4, ITGB1 |
| fca05414 | Dilated cardiomyopathy | LOC102950759, SFXN1, CACNA1D, MYL3, TPM4, ITGB1 |
| fca05144 | Malaria | SFXN1, TLR4, HGF, GYPC |
| fca05014 | Amyotrophic lateral sclerosis (ALS) | GRIA4, LOC102967742, CASP3 |
| fca05016 | Huntington’s disease | CLTCL1, LOC102962163, LOC102957675, CLTC, CASP3, NDUFA4, AP2A2, CREB3L2 |
| fca05162 | Measles | ADAR, TNFAIP3, IFNGR1, TLR4, MAP3K7 |
| fca05168 | Herpes simplex infection | TRAF5, CASP3, MAPK8, MAP3K7, SRSF6, IFNGR1, CCPG1 |
| fca05164 | Influenza A | ADAR, TMPRSS2, TLR4, MAPK8, LOC102967742, IFNGR1 |
| fca05010 | Alzheimer’s disease | YPEL2, ATP2A3, CACNA1D, LOC102953458, CASP3, NDUFA4 |
| fca05012 | Parkinson’s disease | CASP3, NDUFA4, GPR37, LOC102962163, UCHL1 |
| fca05152 | Tuberculosis | TLR4, CASP3, SFXN1, FARSB, MAPK8, IFNGR1 |
| fca05146 | Amoebiasis | SFXN1, TLR4, CASP3 |
| fca05416 | Viral myocarditis | CASP3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, H.; Yu, J.; Li, Q.; Zhang, M. New Evidence of Tiger Subspecies Differentiation and Environmental Adaptation: Comparison of the Whole Genomes of the Amur Tiger and the South China Tiger. Animals 2022, 12, 1817. https://doi.org/10.3390/ani12141817
Du H, Yu J, Li Q, Zhang M. New Evidence of Tiger Subspecies Differentiation and Environmental Adaptation: Comparison of the Whole Genomes of the Amur Tiger and the South China Tiger. Animals. 2022; 12(14):1817. https://doi.org/10.3390/ani12141817
Chicago/Turabian StyleDu, Hairong, Jingjing Yu, Qian Li, and Minghai Zhang. 2022. "New Evidence of Tiger Subspecies Differentiation and Environmental Adaptation: Comparison of the Whole Genomes of the Amur Tiger and the South China Tiger" Animals 12, no. 14: 1817. https://doi.org/10.3390/ani12141817
APA StyleDu, H., Yu, J., Li, Q., & Zhang, M. (2022). New Evidence of Tiger Subspecies Differentiation and Environmental Adaptation: Comparison of the Whole Genomes of the Amur Tiger and the South China Tiger. Animals, 12(14), 1817. https://doi.org/10.3390/ani12141817