
IGF1R and LOX Modules Are Related to Antler Growth Rate Revealed by Integrated Analyses of Genomics and Transcriptomics

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Preparation
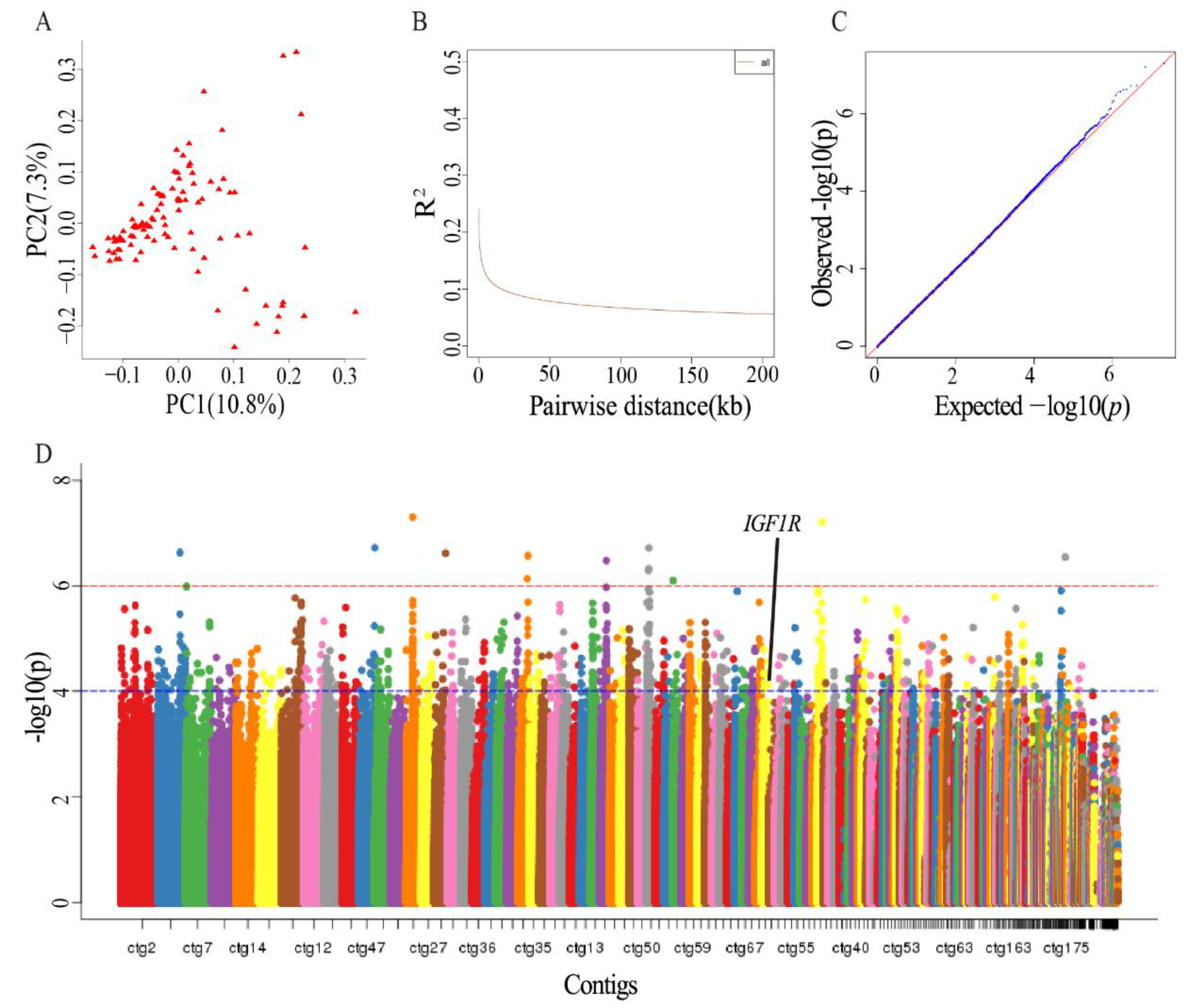
2.2. Identification of Antler-Growth-Rate-Related SNPs Using GWAS
2.3. Identification of Transcripts Related to Antler Growth Rate Using the Integration of GWAS and WGCNA
2.4. Gene Screening in the Modules and the Construction of Gene Interaction Networks
2.5. Verification of Transcripts of Genes Regulating Antler Growth Rate
3. Results
3.1. Identified Genes Related to Antler Growth Rate Using GWAS
3.2. Identified Genes That Are Related to Antler Growth Rate Using the Integration of GWAS and WGCNA
3.3. Functional Enrichment and Interaction Network of Genes Related to Antler Growth Rate
3.4. Expressions of Different Transcripts of IGF1 and IGF1R in Sika Deer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goss, R. Deer Antlers: Regeneration, Function and Evolution; Academic Press: New York, NY, USA, 1983. [Google Scholar]
- Landete-Castillejos, T.; Kierdorf, H.; Gomez, S.; Luna, S.; García, A.J.; Cappelli, J.; Pérez-Serrano, M.; Pérez-Barbería, J.; Gallego, L.; Kierdorf, U. Antlers—Evolution, development, structure, composition, and biomechanics of an outstanding type of bone. Bone 2019, 128, 22. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Li, C.; Liu, Z.; Du, Y. The study on the relationship between antler’s growth rate, relative bone mass and circulating testosterone, estradiol, AKP in sika deer. Acta Vet. Zootech. Sin. 1988, 19, 171–176. [Google Scholar]
- Banks, W.J.; Newbrey, J.W. Light microscopic studies of the ossification process in developing antlers. In Antler Development in Cervidae; Caesar Kleberg Wildlife: Kingsville, TX, USA, 1983; pp. 231–260. [Google Scholar]
- Bubenik, G.A.; Sempere, A.J.; Hamr, J. Developing antler, a model for endocrine regulation of bone growth. Concentration gradient of T3, T4, and alkaline phosphatase in the antler, jugular, and the saphenous veins. Calcif. Tissue Int. 1987, 41, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Clark, D.E.; Lord, E.A.; Stanton, J.A.; Suttie, J.M. Sampling technique to discriminate the different tissue layers of growing antler tips for gene discovery. Anat. Rec. 2002, 268, 125–130. [Google Scholar] [CrossRef]
- Du, Z.-H.; Bai, X.-J. Association analysis between SNPs of the growth hormone gene and antler production in spotted deer. Hereditas (Beijing) 2007, 29, 337–342. [Google Scholar] [CrossRef]
- Xiong, J. Effects of Hormone and Hormone Receptor Gene Mutations on the Regulation of the Antler Production in Sika Deer; Central China Agricultural University: Beijing, China, 2012. [Google Scholar]
- Yang, F.F.; Huo, L.J.; Yang, L.G.; Riaz, H.; Xiong, L.R.; Chen, J.G.; Zhang, S.J.; Xiong, J.J. Association between melatonin receptor 1A (MTNR1A) gene single-nucleotide polymorphisms and the velvet antler yield of Sika deer. Mol. Biol. Rep. 2014, 41, 1901–1906. [Google Scholar] [CrossRef]
- Suttie, J.M.; Gluckman, P.D.; Butler, J.H.; Fennessy, P.F.; Corson, I.D.; Laas, F.J. Insulin-like growth factor 1 (IGF-1) antler-stimulating hormone? Endocrinology 1985, 116, 846–848. [Google Scholar] [CrossRef]
- Suttie, J.M.; Fennessy, P.F.; Gluckman, P.D.; Corson, I.D. Elevated plasma IGF 1 levels in stags prevented from growing antlers. Endocrinology 1988, 122, 3005–3007. [Google Scholar] [CrossRef]
- Hu, P.; Wang, T.; Liu, H.; Xu, J.; Wang, L.; Zhao, P.; Xing, X. Full-length transcriptome and microRNA sequencing reveal the specific gene-regulation network of velvet antler in sika deer with extremely different velvet antler weight. Mol. Genet. Genom. 2019, 294, 431–443. [Google Scholar] [CrossRef]
- Hu, P.F.; Xu, J.P.; Ai, C.; Shao, X.J.; Wang, H.L.; Dong, Y.M.; Cui, X.Z.; Fuhe, Y.; Xiumei, X. Screening weight related genes of velvet antlers by whole genome re-sequencing. Yi Chuan 2017, 39, 1090–1101. [Google Scholar]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farber, C.R. Systems-level analysis of genome-wide association data. G3 2013, 3, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-L.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J.; et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1471–2105. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Ai, C.; Wang, T.; Li, Y.; Liu, H.; Hu, P.; Wang, G.; Liu, H.; Wang, H.; Zhang, R.; et al. The First High-Quality Reference Genome of Sika Deer Provides Insights for High-Tannin Adaptation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Shen, Y.Y.; Lim, B.K.; Liu, H.Q.; Liu, J.; Irwin, D.M.; Zhang, Y.P. Multiple episodes of convergence in genes of the dim light vision pathway in bats. PLoS ONE 2012, 7, 11. [Google Scholar] [CrossRef]
- Li, C.; Harper, A.; Puddick, J.; Wang, W.; McMahon, C. Proteomes and signalling pathways of antler stem cells. PLoS ONE 2012, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhao, H.; Wang, D.; McMahon, C.; Li, C. Differential effects of the PI3K/AKT pathway on antler stem cells for generation and regeneration of antlers in vitro. Front. Biosci. 2018, 23, 1848–1863. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, X.; Shen, X.; Zhang, Y.; Ye, L.; Li, D.; Zhu, Q.; Yin, H. CircCCDC91 regulates chicken skeletal muscle development by sponging miR-15 family via activating IGF1-PI3K/AKT signaling pathway. Poult. Sci. 2022, 101, 25. [Google Scholar] [CrossRef]
- Elliott, J.L.; Oldham, J.M.; Ambler, G.R.; Bass, J.J.; Spencer, G.S.; Hodgkinson, S.C.; Breier, B.H.; Gluckman, P.D.; Suttie, J.M. Presence of insulin-like growth factor-I receptors and absence of growth hormone receptors in the antler tip. Endocrinology 1992, 130, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.L.; Oldham, J.M.; Ambler, G.R.; Molan, P.C.; Spencer, G.S.; Hodgkinson, S.C.; Breier, B.H.; Gluckman, P.D.; Suttie, J.M.; Bass, J.J. Receptors for insulin-like growth factor-II in the growing tip of the deer antler. J. Endocrinol. 1993, 138, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Price, J.S.; Oyajobi, B.O.; Oreffo, R.O.; Russell, R.G. Cells cultured from the growing tip of red deer antler express alkaline phosphatase and proliferate in response to insulin-like growth factor-I. J. Endocrinol. 1994, 143, R9–R16. [Google Scholar] [CrossRef] [PubMed]
- Sadighi, M.; Haines, S.R.; Skottner, A.; Harris, A.J.; Suttie, J.M. Effects of insulin-like growth factor-I (IGF-I) and IGF-II on the growth of antler cells in vitro. J. Endocrinol. 1994, 143, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Littlejohn, R.P.; Suttie, J.M. Effects of insulin-like growth factor 1 and testosterone on the proliferation of antlerogenic cells in vitro. J. Exp. Zool. 1999, 284, 82–90. [Google Scholar] [CrossRef]
- Zhou, W.; Rowitz, B.M.; Dailey, M.J. Insulin/IGF-1 enhances intestinal epithelial crypt proliferation through PI3K/Akt, and not ERK signaling in obese humans. Exp. Biol. Med. 2018, 243, 911–916. [Google Scholar] [CrossRef]
- Fu, S.; Yin, L.; Lin, X.; Lu, J.; Wang, X. Effects of Cyclic Mechanical Stretch on the Proliferation of L6 Myoblasts and Its Mechanisms: PI3K/Akt and MAPK Signal Pathways Regulated by IGF-1 Receptor. Int. J. Mol. Sci. 2018, 19, 1649. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, D.; Zhang, Y.; Ni, N.; Tang, Z.; Bai, Z.; Shen, B.; Sun, H.; Gu, P. Insulin-like growth factor-1 regulation of retinal progenitor cell proliferation and differentiation. Cell Cycle 2018, 17, 515–526. [Google Scholar] [CrossRef]
- Ayub, A.; Yip, W.K.; Seow, H.F. Dual treatments targeting IGF-1R, PI3K, mTORC or MEK synergize to inhibit cell growth, induce apoptosis, and arrest cell cycle at G1 phase in MDA-MB-231 cell line. Biomed. Pharmacother. 2015, 75, 40–50. [Google Scholar] [CrossRef]
- Slomiany, M.G.; Black, L.A.; Kibbey, M.M.; Day, T.A.; Rosenzweig, S.A. IGF-1 induced vascular endothelial growth factor secretion in head and neck squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2006, 342, 851–858. [Google Scholar] [CrossRef]
- Parry, A.M.; Price, J.S.; Bentley, H.; Russell, R.G.G. P51. Transforming growth factor-beta expression in regenerating deer antler. Bone 1996, 19, 699. [Google Scholar] [CrossRef]
- Francis, S.; Suttie, J. Detection of growth factors and proto-oncogene mRNA in the growing tip of red deer (Cervus elaphus) antler using reverse-transcriptase polymerase chain reaction (RT-PCR). J. Exp. Zool. 1998, 281, 36–42. [Google Scholar] [CrossRef]
- Barling, P.M.; Lai, A.K.; Nicholson, L.F. Distribution of EGF and its receptor in growing red deer antler. Cell Biol. Int. 2005, 29, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Lord, E.A.; Suttie, J.M. Expression of VEGF and pleiotrophin in deer antler. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2006, 288, 1281–1293. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Genes |
|---|---|
| vascular endothelial growth factor receptor signaling pathway | NRP2, DOCK1, NRP1, BAIAP2, RHOA, PIK3R1, PXN, NCKAP1 |
| epidermal growth factor receptor signaling pathway | SHC1, PIK3R1, IQGAP1, PXN, PTPN11 |
| ERBB2 (HER2) signaling pathway | UBC, SHC1, PIK3R1, CDC37 |
| cellular response to epidermal growth factor stimulus | MCM7, COL1A1, SNAI2, IQGAP1 |
| insulin receptor signaling pathway | IGF1R, BAIAP2, SHC1, APPL1, PIK3R1 |
| platelet-derived growth factor receptor signaling pathway | NRP1, IQGAP1, PTPN11 |
| transforming growth factor beta receptor signaling pathway | COL3A1, RHOA, COL1A2, UBC, ITGB5, ENG, PXN |
| negative regulation of transforming growth factor beta receptor signaling pathway | GLG1, HTRA1, UBC, ENG |
| ephrin receptor (subfamily of RTKs) signaling pathway | ARPC1A, ARPC1B, AP2A2, ARPC2, RHOA, SDC2, PTPN11 |
| activation of MAPK activity | CD81, UBC, SHC1, LPAR1, PTPN11 |
| positive regulation of mesenchymal cell proliferation | FGFR2, PRRX1, CTNNB1 |
| PI3K-Akt signaling pathway | HSP90AB1, FGFR2, COL3A1, ITGA11, ITGB5, LPAR1, CDC37, IGF1R, CCND1, HSP90B1, LAMA4, COMP, COL1A2, YWHAQ, COL6A1, TNN, NOS3, GNB4, COL1A1, LAMC1, COL11A2, THBS2, COL11A1, PIK3R1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, P.; Wang, Z.; Li, J.; Wang, D.; Wang, Y.; Zhao, Q.; Li, C. IGF1R and LOX Modules Are Related to Antler Growth Rate Revealed by Integrated Analyses of Genomics and Transcriptomics. Animals 2022, 12, 1522. https://doi.org/10.3390/ani12121522
Hu P, Wang Z, Li J, Wang D, Wang Y, Zhao Q, Li C. IGF1R and LOX Modules Are Related to Antler Growth Rate Revealed by Integrated Analyses of Genomics and Transcriptomics. Animals. 2022; 12(12):1522. https://doi.org/10.3390/ani12121522
Chicago/Turabian StyleHu, Pengfei, Zhen Wang, Jiping Li, Dongxu Wang, Yusu Wang, Quanmin Zhao, and Chunyi Li. 2022. "IGF1R and LOX Modules Are Related to Antler Growth Rate Revealed by Integrated Analyses of Genomics and Transcriptomics" Animals 12, no. 12: 1522. https://doi.org/10.3390/ani12121522
APA StyleHu, P., Wang, Z., Li, J., Wang, D., Wang, Y., Zhao, Q., & Li, C. (2022). IGF1R and LOX Modules Are Related to Antler Growth Rate Revealed by Integrated Analyses of Genomics and Transcriptomics. Animals, 12(12), 1522. https://doi.org/10.3390/ani12121522