
Genomic Population Structure of the Main Historical Genetic Lines of Spanish Merino Sheep

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Sampling
2.2. Genotyping and Quality Control
2.3. Construction of a Worldwide Merino Breeds Database
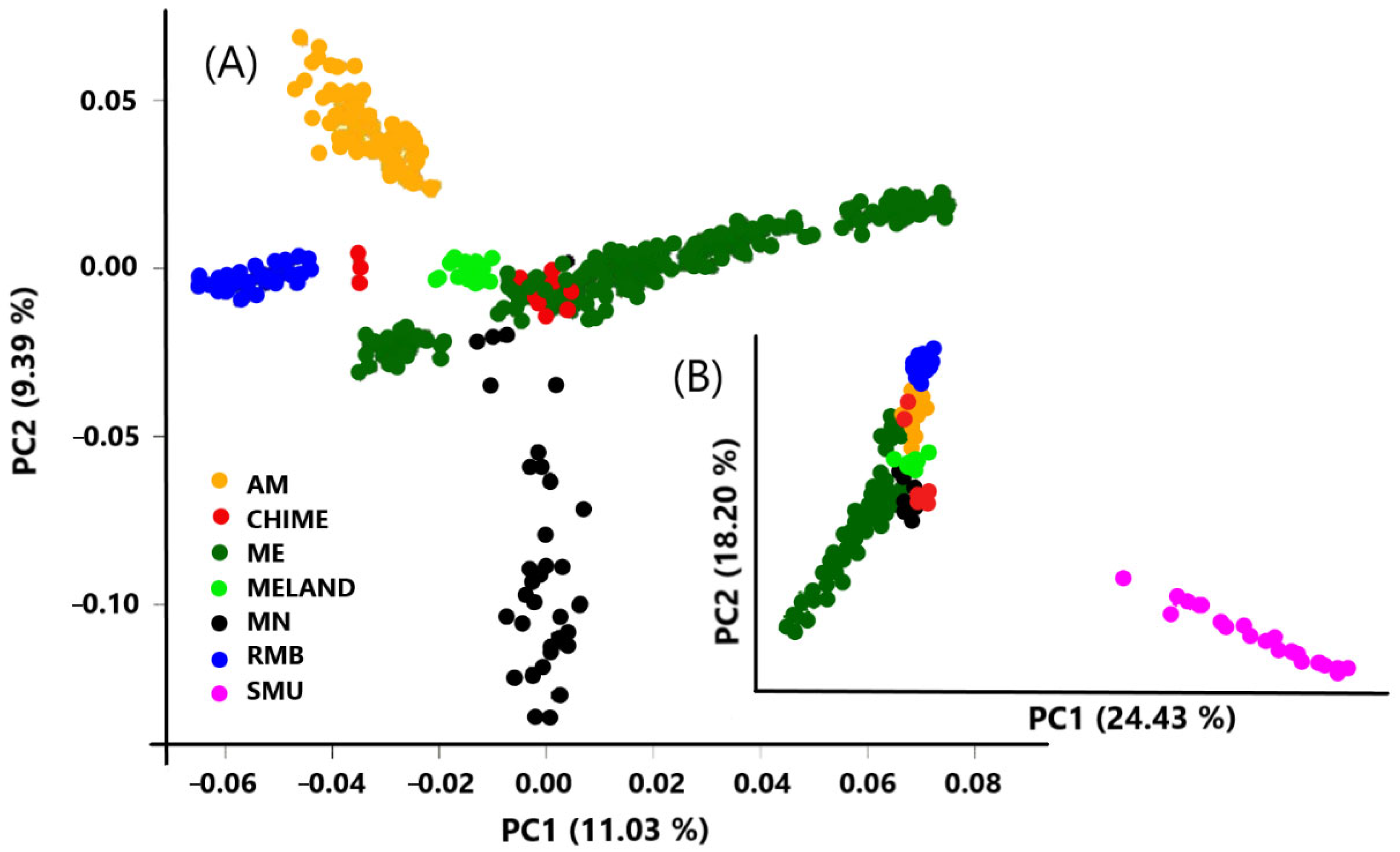
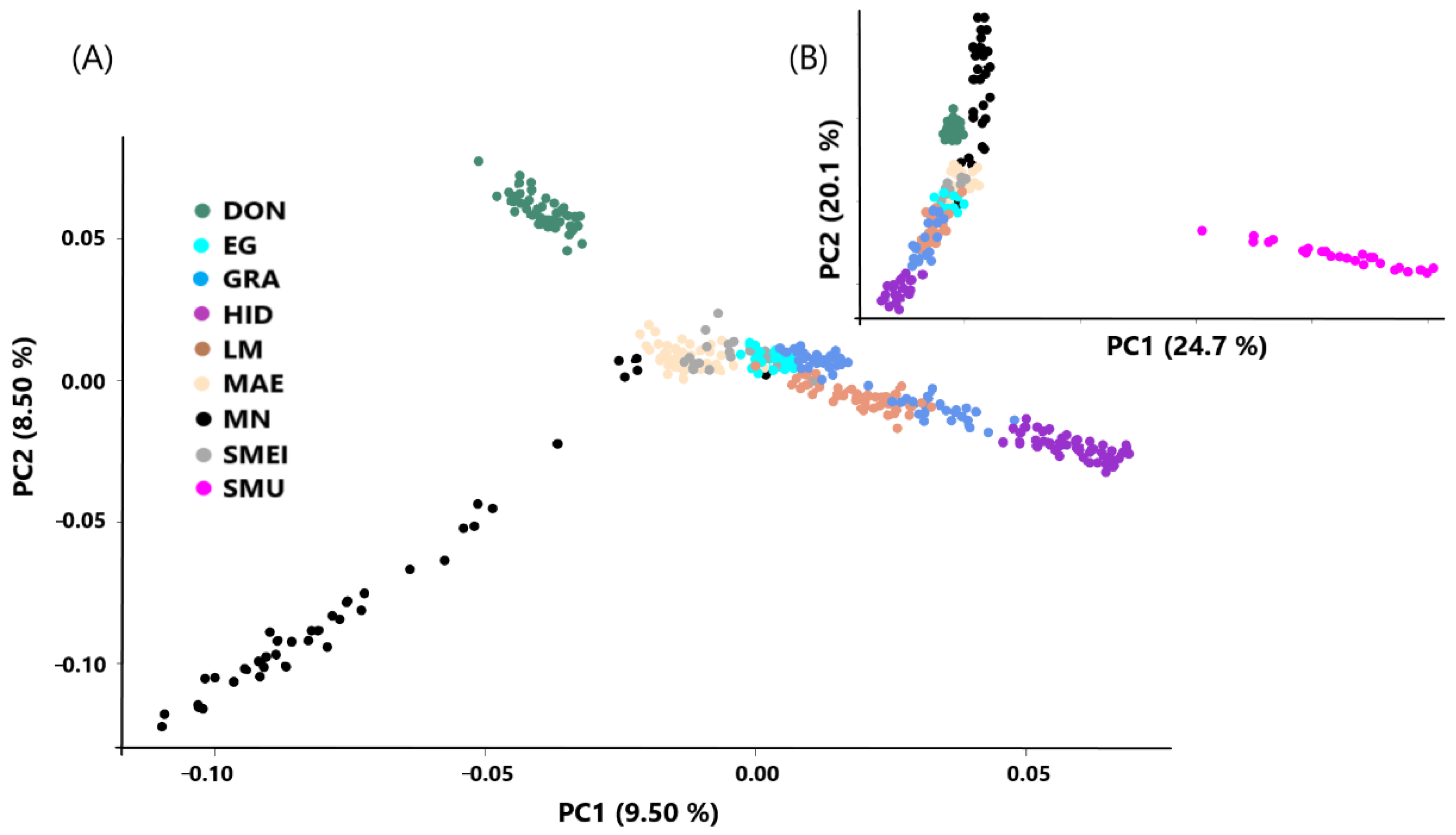
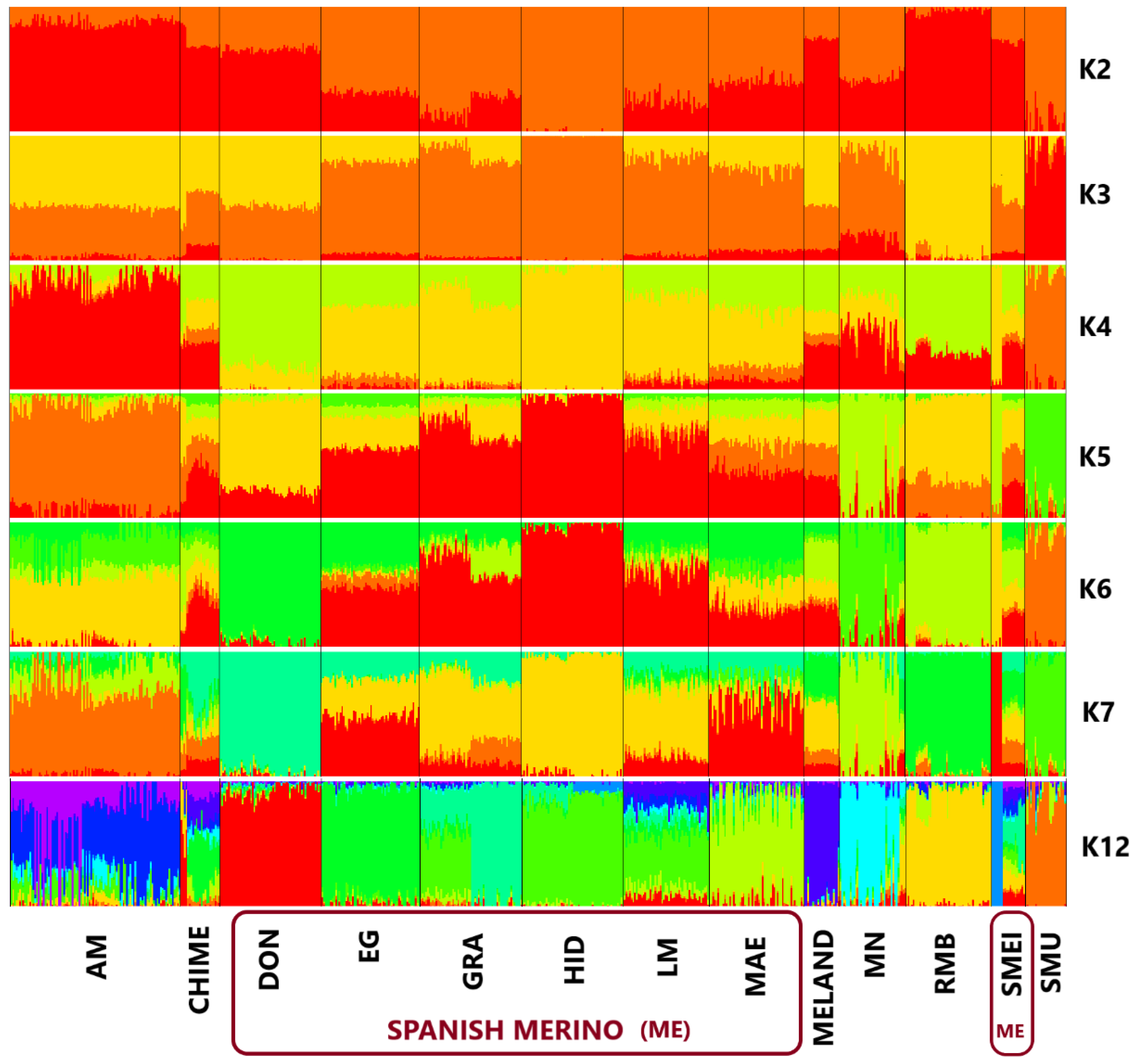
2.4. Population Structure and Genomic Differentiation
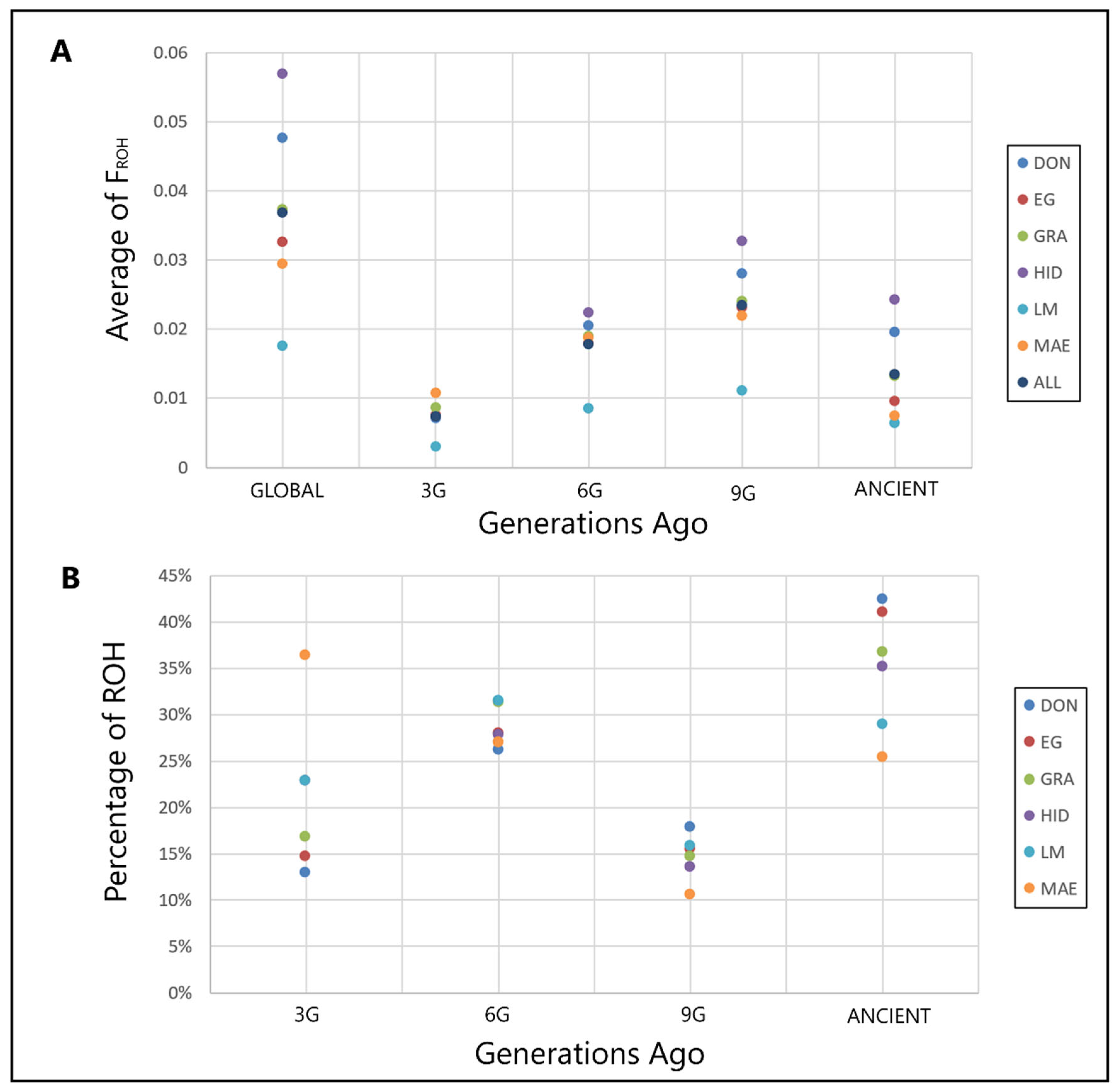
2.5. Analysis of ROH Patterns
2.6. Analysis of the Molecular Variance (AMOVA)
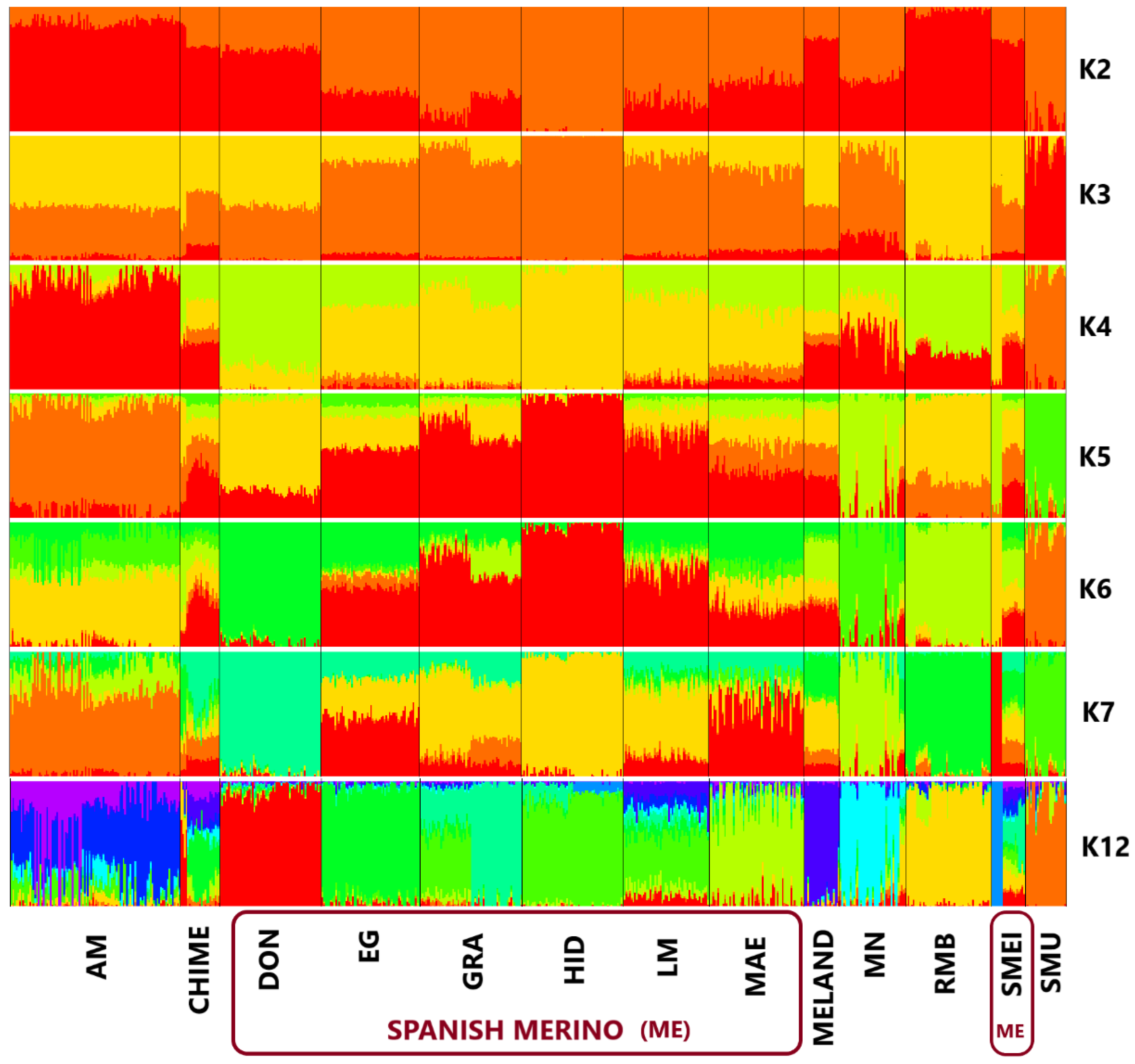
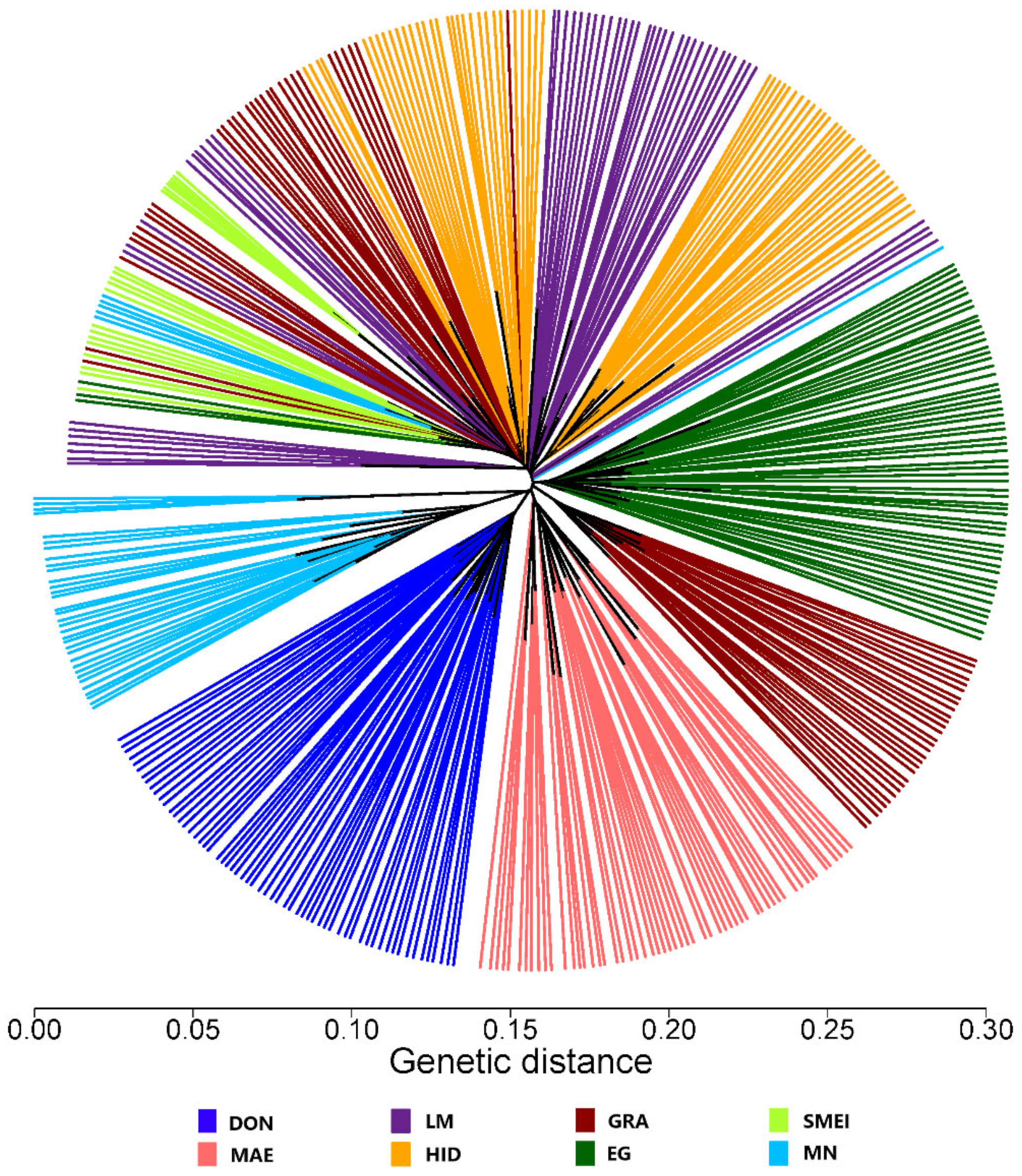
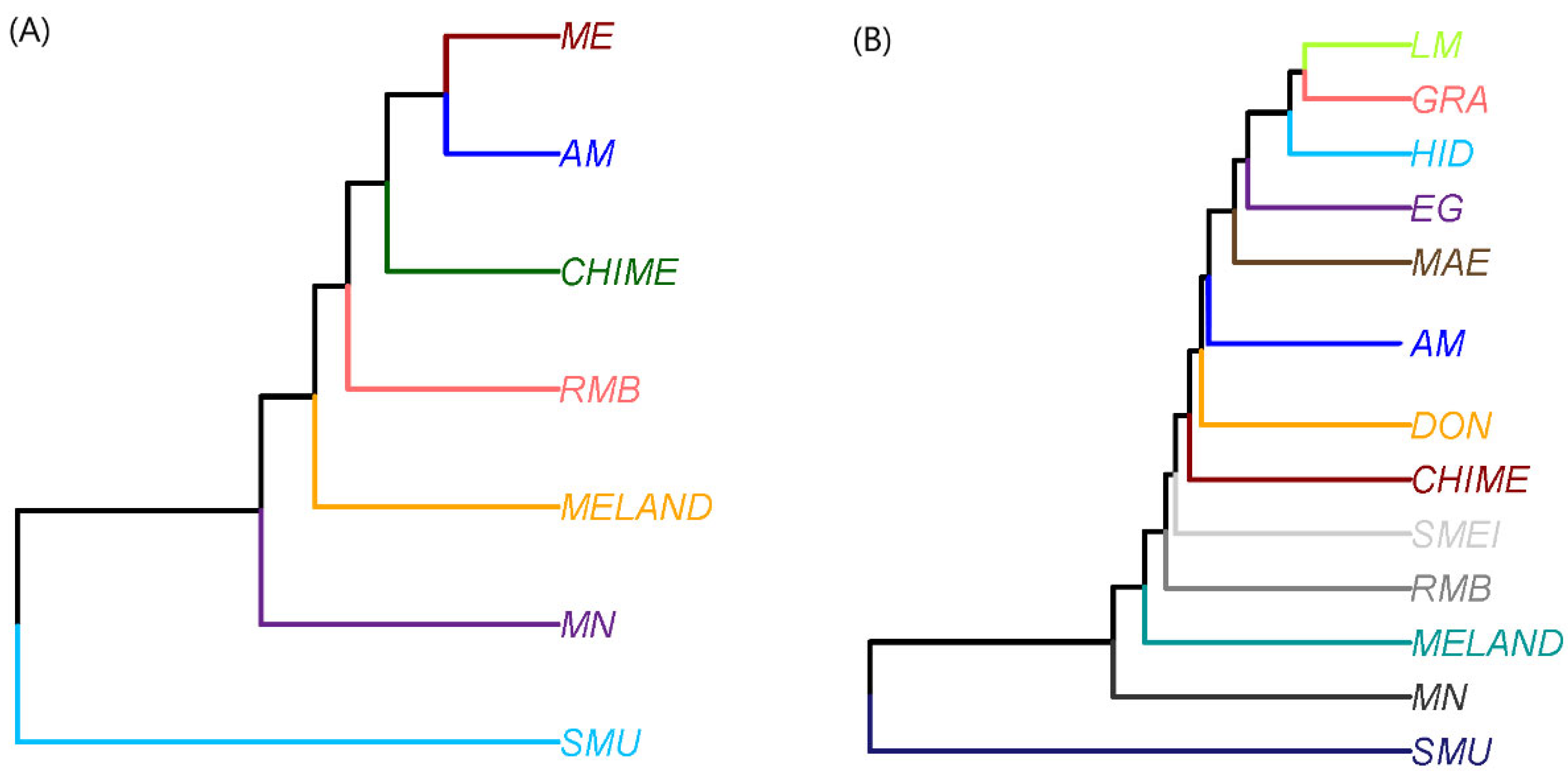
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MAPA. Sistema Nacional de Información de Razas (ARCA). Available online: https://www.mapa.gob.es/es/ganaderia/temas/zootecnia/razas-ganaderas/razas/catalogo-razas/ovino/merina/iframe-ejemplo-arca.aspx (accessed on 2 March 2022).
- Fuentes, F.C.; Sánchez, J.M.; Gonzalo, C. Manual de Etnología Animal: Razas de Rumiantes; Diego Marín: Murcia, Spain, 2000; p. 496. [Google Scholar]
- Ryder, M.L. Sheep and Man; Duckworth: London, UK, 1983; p. 856. [Google Scholar]
- Sánchez-Belda, A.; Sanchez-Trujillano, M. Razas Ovinas Españolas; Ministerio de Agricultura, Spain: Madrid, Spain, 1986; p. 887.
- Cano, T.P.; Molina, A. Las razas ovinas andaluzas de fomento merina y segureña. In Las Razas Ganaderas de Andalucía. Vol. Ii, Patrimonio Ganadero Andaluz; Junta de Andalucía. Consejería de Agricultura y Pesca: Sevilla, Spain, 2007; Volume 2, pp. 260–300. [Google Scholar]
- Sánchez, G.A. De etnología ovina. Merinos. A propósito de dos testas de carnero esculpidas en el sarcófago romano de córdoba. Archivos de zootecnia 1970, 19, 375–390. [Google Scholar]
- Zorita, E. Hacia una nueva estructura de la ganadería ovina en españa, armonizando recursos alimenticios y objetivos medioambientales. Ovis 1990, 11, 9–42. [Google Scholar]
- Garzón, R.; Luque, J.; Llanes, D.; Povedano, C.; Rodero, A.; Rodero, J.; Vallejo, M.; Zarazaga, I. Fundamentos Históricos y Genéticos del Merino Español; Publicaciones del Monte de Piedad y Caja de Ahorros de Córdoba: Cordoba, Spain, 1977; p. 145. [Google Scholar]
- Laguna, E. Historia del Merino; Ministerio de Agricultura, Spain: Madrid, Spain, 1986; p. 224.
- Muñoz, C.E. El ovino en américa. Participación del merino en la formación de la cabaña americana. In Cincuentenario de la Asociación del Cuerpo Nacional de Veterinarios; Secretaría General Técnica: Madrid, Spain, 2004; pp. 253–265. [Google Scholar]
- Arrebola, F.V.M.; Molina, A.; Barajas, F. Selección para caracteres laneros en merino autóctono español. Feagas 2003, 24, 95–99. [Google Scholar]
- Mason, I.L. Mediterranean breeds of cattle, sheep and goats. In Isotope Aided Studies on Livestock Productivity in Mediterranean and North African Countries; IAAE: Rotterdam, The Netherlands, 1988; p. 189. [Google Scholar]
- Ciani, E.; Lasagna, E.; D’Andrea, M.; Alloggio, I.; Marroni, F.; Ceccobelli, S.; Delgado Bermejo, J.V.; Sarti, F.M.; Kijas, J.; Lenstra, J.A.; et al. Merino and merino-derived sheep breeds: A genome-wide intercontinental study. Genet. Sel. Evol. 2015, 47, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, R.; Orel, V. Genetic Prehistory in Selective Breeding: A Prelude to Mendel; Cambridge University Press: Cambridge, MA, USA, 2003; p. 342. [Google Scholar]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [Green Version]
- Rochus, C.M.; Tortereau, F.; Plisson-Petit, F.; Restoux, G.; Moreno-Romieux, C.; Tosser-Klopp, G.; Servin, B. Revealing the selection history of adaptive loci using genome-wide scans for selection: An example from domestic sheep. BMC Genom. 2018, 19, 71. [Google Scholar] [CrossRef] [Green Version]
- Fésüs, L.S.L.; Hajduk, P.; Székely, P. The determining role of merino in hungarian sheep breeding. In Proceedings of the 6th Merino World Conference, Budapest, Hungary, 29 April–1 May 2002. [Google Scholar]
- Valera, M.; Arrebola, F.; Juárez, M.; Molina, A. Genetic improvement of wool production in spanish merino sheep: Genetic parameters and simulation of selection strategies. Anim. Prod. Sci. 2009, 49, 43–47. [Google Scholar] [CrossRef]
- Esteban, M. La Raza Merina y Sus Cruces en la Producción de Carne; Ministerio de Agricultura Pesca y Alimentación del Gobierno de España: Madrid, Spain, 1994; p. 175.
- Peña Blanco, F.; Alcalde Aldea, M.J. Las razas ovinas integradas en andalucía: Merino precoz francés, ille de france, fleischscaff, landschaff y lacaune. In Las Razas Ganaderas de Andalucía. Patrimonio Ganadero Patrimonio Ganadero Andaluz; Junta de Andalucia, Spain: Andalusia, Spain, 2007; Volume 2, pp. 331–364. [Google Scholar]
- Azor, P.J.; Cervantes, I.; Valera, M.; Arranz, J.J.; Medina, C.; Gutiérrez, J.P.; Goyache, F.; Muñoz, A.; Molina, A. Preliminary assessment of population structure of spanish merino breed: Traditional strains situation using genealogical and molecular analysis. ITEA Inf. Tecn. Econ. Agrar. 2008, 104, 295–302. [Google Scholar]
- Granero, A.M.; Molina, A.; Anaya, G.; Ziadib, C.; Alcalde, M.J. Morphometric differences based on quantitative traits between different genetic lines in the merino español sheep breed. Aspa 24th congress book of abstract. Ital. J. Anim. Sci. 2021, 20. [Google Scholar] [CrossRef]
- Rodríguez Pascual, M. La Trashumancia. Cultura, Cañadas y Viajes; Edilesa: Leon, Spain, 2001; p. 460. [Google Scholar]
- Lenstra, J.; Groeneveld, L.; Eding, H.; Kantanen, J.; Williams, J.; Taberlet, P.; Nicolazzi, E.; Sölkner, J.; Simianer, H.; Ciani, E.; et al. Molecular tools and analytical approaches for the characterization of farm animal genetic diversity. Anim. Genet. 2012, 43, 483–502. [Google Scholar] [CrossRef]
- Gill, P. An assessment of the utility of single nucleotide polymorphisms (snps) for forensic purposes. Int. J. Legal Med. 2001, 114, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Paschou, P.; Ziv, E.; Burchard, E.G.; Choudhry, S.; Rodriguez-Cintron, W.; Mahoney, M.W.; Drineas, P. Pca-correlated snps for structure identification in worldwide human populations. PLoS Genet. 2007, 3, 1672–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciani, E.; Crepaldi, P.; Nicoloso, L.; Lasagna, E.; Sarti, F.M.; Moioli, B.; Napolitano, F.; Carta, A.; Usai, G.; D’Andrea, M.; et al. Genome-wide analysis of italian sheep diversity reveals a strong geographic pattern and cryptic relationships between breeds. Anim. Genet. 2014, 45, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mousel, M.R.; Wu, X.; Michal, J.J.; Zhou, X.; Ding, B.; Dodson, M.V.; El-Halawany, N.K.; Lewis, G.S.; Jiang, Z. Genome-wide genetic diversity and differentially selected regions among suffolk, rambouillet, columbia, polypay, and targhee sheep. PLoS ONE 2013, 8, e65942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniskova, T.E.; Dotsev, A.V.; Selionova, M.I.; Kunz, E.; Medugorac, I.; Reyer, H.; Wimmers, K.; Barbato, M.; Traspov, A.A.; Brem, G.; et al. Population structure and genetic diversity of 25 russian sheep breeds based on whole-genome genotyping. Genet. Sel. Evol. 2018, 50, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, A.C.; Purfield, D.C.; Judge, M.M.; Long, C.; Fair, S.; Berry, D.P. Population structure and breed composition prediction in a multi-breed sheep population using genome-wide single nucleotide polymorphism genotypes. Animal 2020, 14, 464–474. [Google Scholar] [CrossRef]
- Ben Jemaa, S.; Kdidi, S.; Gdura, A.M.; Dayhum, A.S.; Eldaghayes, I.M.; Boussaha, M.; Rebours, E.; Yahyaoui, M.H. Inferring the population structure of the maghreb sheep breeds using a medium-density snp chip. Anim. Genet. 2019, 50, 526–533. [Google Scholar] [CrossRef]
- Edea, Z.; Dessie, T.; Dadi, H.; Do, K.-T.; Kim, K.-S. Genetic diversity and population structure of ethiopian sheep populations revealed by high-density snp markers. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- FAO. 11th Session of the Itwg on Animal Genetic Resources for Food and Agriculture. Available online: http://www.fao.org/animal-genetics/events/events-detail/en/c/1369166/ (accessed on 2 March 2022).
- Thermofisher. Axiom Cnv Summary Tool User Manual. Available online: https://tools.thermofisher.com/content/sfs/manuals/axiom_cnv_summary_tool_usermanual.pdf (accessed on 15 August 2020).
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation plink: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4. [Google Scholar] [CrossRef]
- R-Core-Team. R: A Language and Environment for Statistical Computing V4.1.3 "One Push Up"; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Gruber, B.; Unmack, P.J.; Berry, O.F.; Georges, A. Dartr: An r package to facilitate analysis of snp data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2018, 18, 691–699. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in r. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.R.; Kadri, N.K.; Flori, L.; Gautier, M.; Druet, T. Rzooroh: An r package to characterize individual genomic autozygosity and identify homozygous-by-descent segments. Methods Ecol. Evol. 2019, 10, 860–866. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using plink? A guide for analyzing medium density snp data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. Detectruns: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes in R. 2018. Available online: https://github.com/bioinformatics-ptp/detectRUNS/tree/master/detectRUNS (accessed on 2 March 2022).
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in european populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.A. A fuller theory of junctions in inbreeding. Heredity 1954, 8, 187–197. [Google Scholar] [CrossRef]
- Goszczynski, D.; Molina, A.; Terán, E.; Morales-Durand, H.; Ross, P.; Cheng, H.; Giovambattista, G.; Demyda-Peyrás, S. Runs of homozygosity in a selected cattle population with extremely inbred bulls: Descriptive and functional analyses revealed highly variable patterns. PLoS ONE 2018, 13, e0200069. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Gosselin, T. Radiator: Radseq Data Exploration, Manipulation and Visualization Using R. V1.1.8. 2020. Available online: https://rdrr.io/github/thierrygosselin/radiator/f/vignettes/rad_genomics_computer_setup.Rmd (accessed on 2 March 2022).
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel r tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef] [Green Version]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing genomic information for livestock improvement. Nat. Rev. Gen. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Groeneveld, L.F.; Lenstra, J.A.; Eding, H.; Toro, M.A.; Scherf, B.; Pilling, D.; Negrini, R.; Finlay, E.K.; Jianlin, H.; Groeneveld, E.; et al. Genetic diversity in farm animals—A review. Anim. Genet. 2010, 41, 6–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawęcka, A.; Gurgul, A.; Miksza-Cybulska, A. The use of snp microarrays for biodiversity studies of sheep: A review. Annals of Animal Science 2016, 16, 975–987. [Google Scholar] [CrossRef] [Green Version]
- Arranz, J.J.; Bayón, Y.; Primitivo, F.S. Genetic relationships among spanish sheep using microsatellites. Anim. Genet. 1998, 29, 435–440. [Google Scholar] [CrossRef]
- Diez-Tascón, C.; Littlejohn, R.P.; Almeida, P.A.; Crawford, A.M. Genetic variation within the merino sheep breed: Analysis of closely related populations using microsatellites. Anim. Genet. 2000, 31, 243–251. [Google Scholar] [CrossRef]
- Poplin, F. Origin of the corsican mouflon in a new paleontologic prospect: By feralization (in french). Ann. Genet. Sel. Anim. 1979, 11, 133–143. [Google Scholar] [PubMed]
- Barbato, M.; Hailer, F.; Orozco-terWengel, P.; Kijas, J.; Mereu, P.; Cabras, P.; Mazza, R.; Pirastru, M.; Bruford, M.W. Genomic signatures of adaptive introgression from european mouflon into domestic sheep. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigne, J.D. Zooarchaeology and the biogeographical history of the mammals of corsica and sardinia since the last ice age. Mammal Rev. 1992, 22, 87–96. [Google Scholar] [CrossRef]
- FAO. Secondary Guidelines for Development of National Farm Animal Genetic Resources Management Plans—New Microsatellite Marker sets; FAO: Rome, Italy, 1993. [Google Scholar]
- Fernández, M.E.; Goszczynski, D.E.; Lirón, J.P.; Villegas-Castagnasso, E.E.; Carino, M.H.; Ripoli, M.V.; Rogberg-Muñoz, A.; Posik, D.M.; Peral-García, P.; Giovambattista, G. Comparison of the effectiveness of microsatellites and snp panels for genetic identification, traceability and assessment of parentage in an inbred angus herd. Genet. Mol. Biol. 2013, 36, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, K.; Panigrahi, M.; Kumar, H.; Bhushan, B.; Dutt, T.; Mishra, B. Genome-wide analysis of genetic diversity and selection signatures in three indian sheep breeds. Livest. Sci. 2021, 243, 104367. [Google Scholar] [CrossRef]
- Nel, C.; Gurman, P.; Swan, A.; van der Werf, J.; Snyman, M.; Dzama, K.; Gore, K.; Scholtz, A.; Cloete, S. The genomic structure of isolation across breed, country and strain for important south african and australian sheep populations. BMC Genom. 2022, 23, 23. [Google Scholar] [CrossRef]
- Bedhiaf-Romdhani, S.; Baazaoui, I.; Ciani, E.; Mastrangelo, S.; Sassi, M.B. Genetic structure of tunisian sheep breeds as inferred from genome-wide snp markers. Small Rumin. Res. 2020, 191. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Gen. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guo, J.; Li, F.; Niu, C. Evaluation of crossbreeding of australian superfine merinos with gansu alpine finewool sheep to improve wool characteristics. PLoS ONE 2016, 11, e0166374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
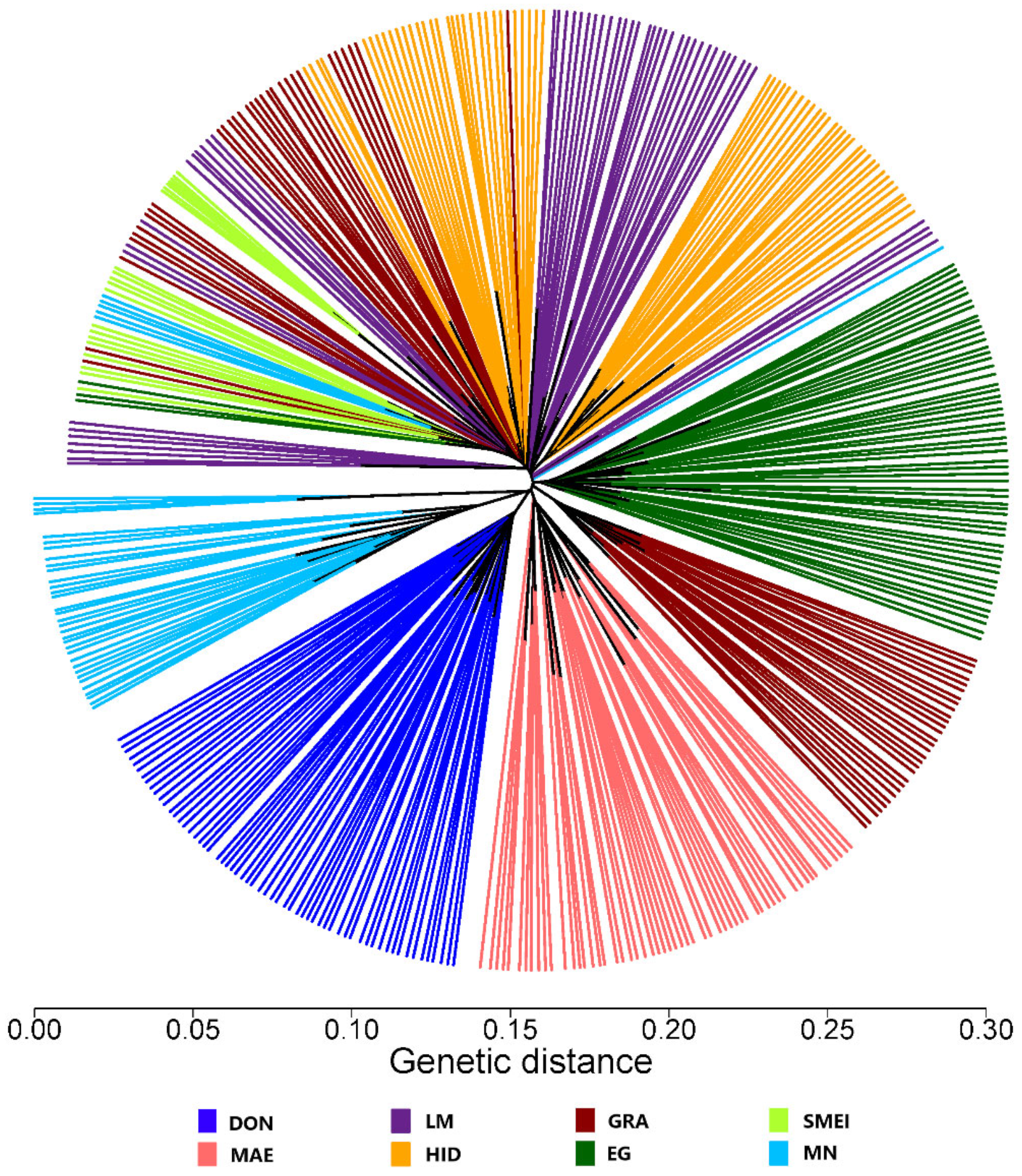


{kind=link}
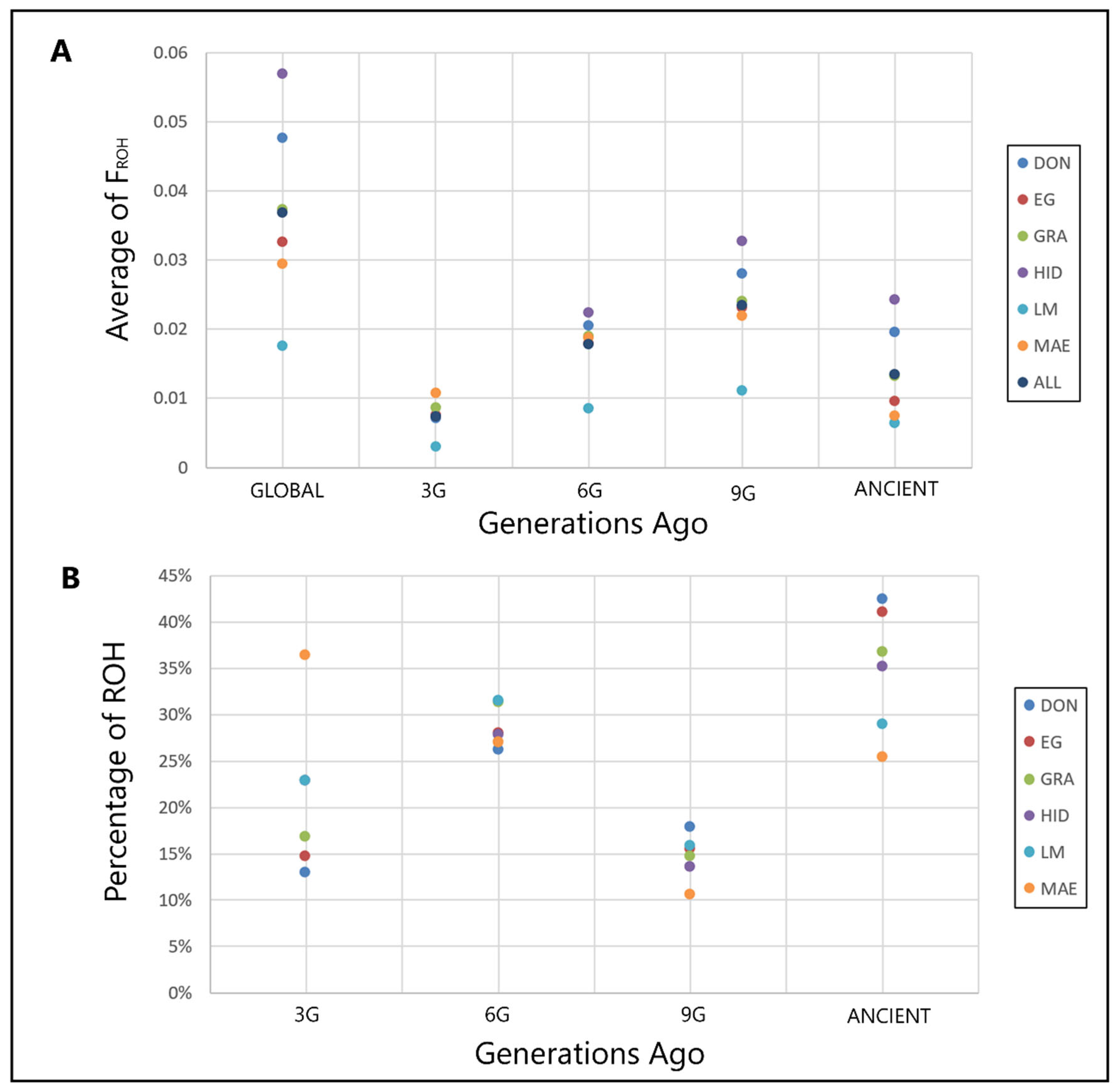
{kind=link}
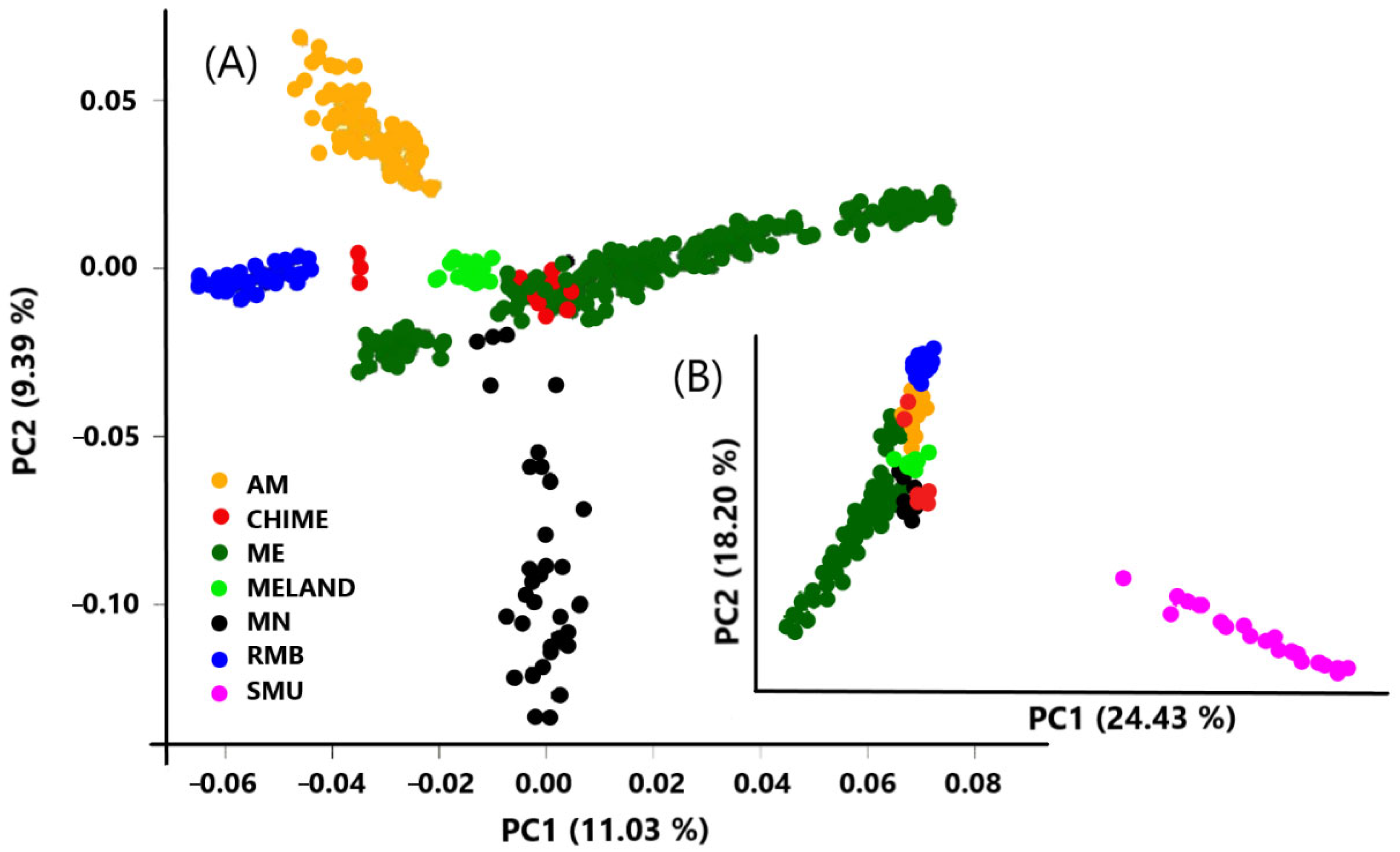
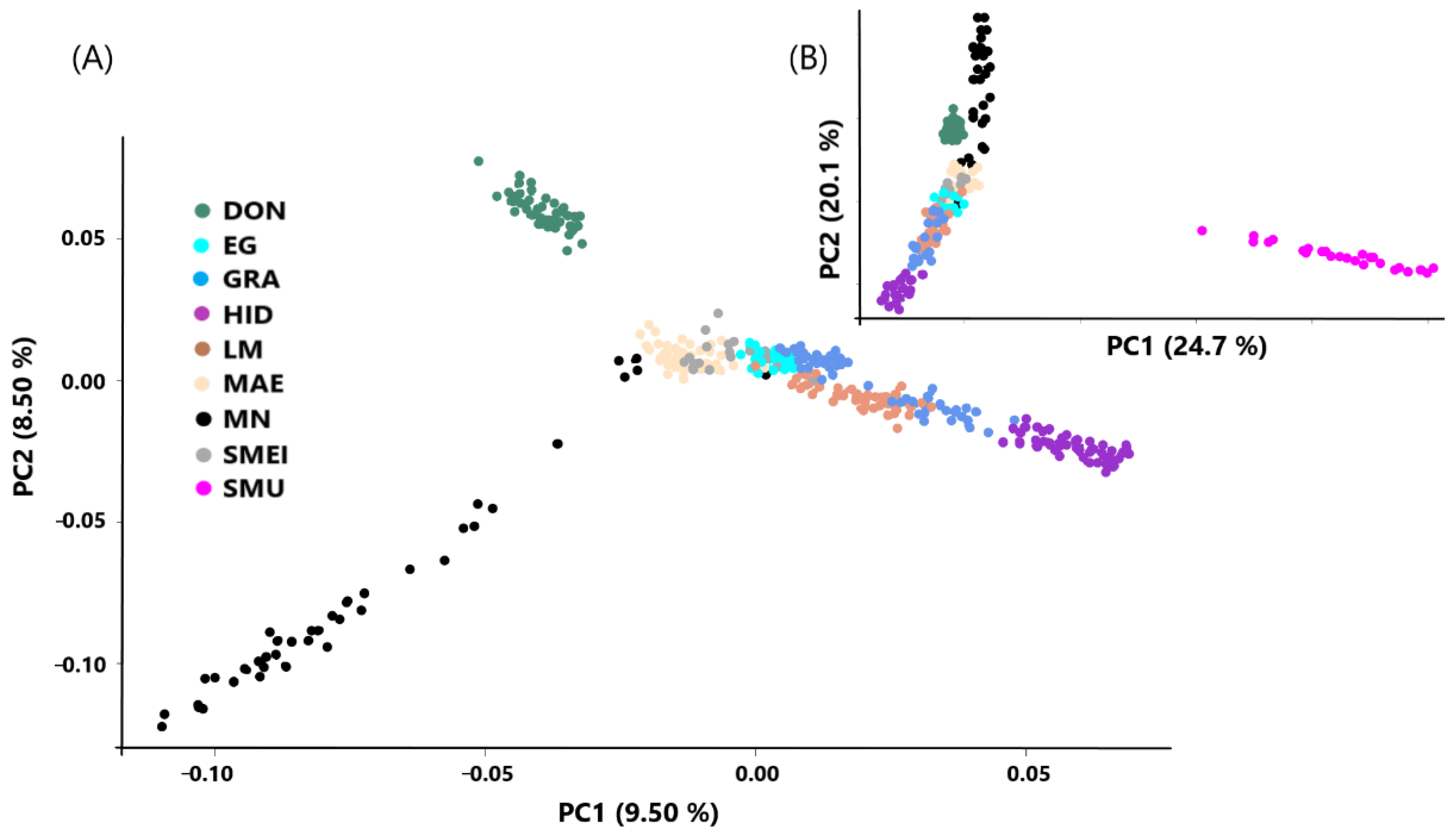{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Breed | N | Ho | He |
|---|---|---|---|---|
| AM | Australian Merino | 100 | 0.361 | 0.375 |
| CHIME | Chinese Merino | 23 | 0.358 | 0.367 |
| MELAND | Landschaf Merino | 20 | 0.358 | 0.356 |
| RMB | Rambouillet Merino | 50 | 0.341 | 0.356 |
| ME | Whole White Spanish Merino | 364 | 0.363 | 0.374 |
| DON | Spanish Merino | 60 | 0.359 | 0.357 |
| EG | Spanish Merino | 58 | 0.367 | 0.364 |
| GRA | Spanish Merino | 60 | 0.366 | 0.367 |
| HID | Spanish Merino | 60 | 0.355 | 0.357 |
| MAE | Spanish Merino | 56 | 0.369 | 0.366 |
| LM | Spanish Merino | 50 | 0.375 | 0.372 |
| SMEI | Spanish Merino | 20 | 0.331 | 0.358 |
| MN | Spanish Black Merino | 39 | 0.358 | 0.349 |
| SMU | Mouflon | 24 | 0.334 | 0.312 |
| N | N Pop | % of Variation | |||
|---|---|---|---|---|---|
| A | B | C | |||
| Merino derived Breeds 1 | 620 | 6 | 5.01 | 2.75 | 92.23 |
| Spanish Merino genetic lines 2 | 364 | 7 | 3.99 | 0.12 | 95.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granero, A.; Anaya, G.; Demyda-Peyrás, S.; Alcalde, M.J.; Arrebola, F.; Molina, A. Genomic Population Structure of the Main Historical Genetic Lines of Spanish Merino Sheep. Animals 2022, 12, 1327. https://doi.org/10.3390/ani12101327
Granero A, Anaya G, Demyda-Peyrás S, Alcalde MJ, Arrebola F, Molina A. Genomic Population Structure of the Main Historical Genetic Lines of Spanish Merino Sheep. Animals. 2022; 12(10):1327. https://doi.org/10.3390/ani12101327
Chicago/Turabian StyleGranero, Antonio, Gabriel Anaya, Sebastián Demyda-Peyrás, María J. Alcalde, Francisco Arrebola, and Antonio Molina. 2022. "Genomic Population Structure of the Main Historical Genetic Lines of Spanish Merino Sheep" Animals 12, no. 10: 1327. https://doi.org/10.3390/ani12101327
APA StyleGranero, A., Anaya, G., Demyda-Peyrás, S., Alcalde, M. J., Arrebola, F., & Molina, A. (2022). Genomic Population Structure of the Main Historical Genetic Lines of Spanish Merino Sheep. Animals, 12(10), 1327. https://doi.org/10.3390/ani12101327