
Microbial Diversity of the Chinese Tiger Frog (Hoplobatrachus rugulosus) on Healthy versus Ulcerated Skin


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Amplification, and Sequencing
2.3. Data Analysis
3. Results
3.1. Sequencing Data
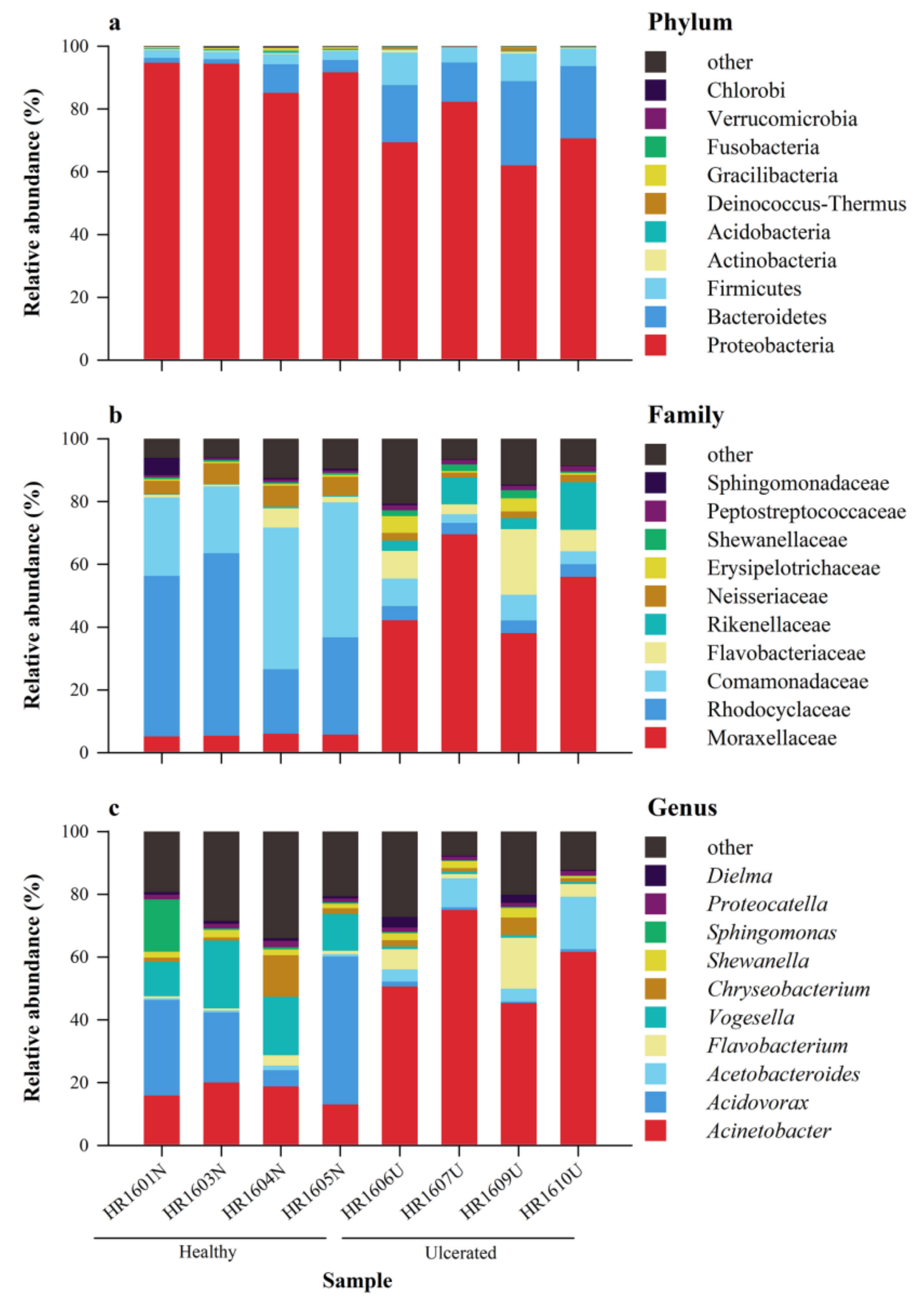
3.2. Composition of the Skin Microbiota
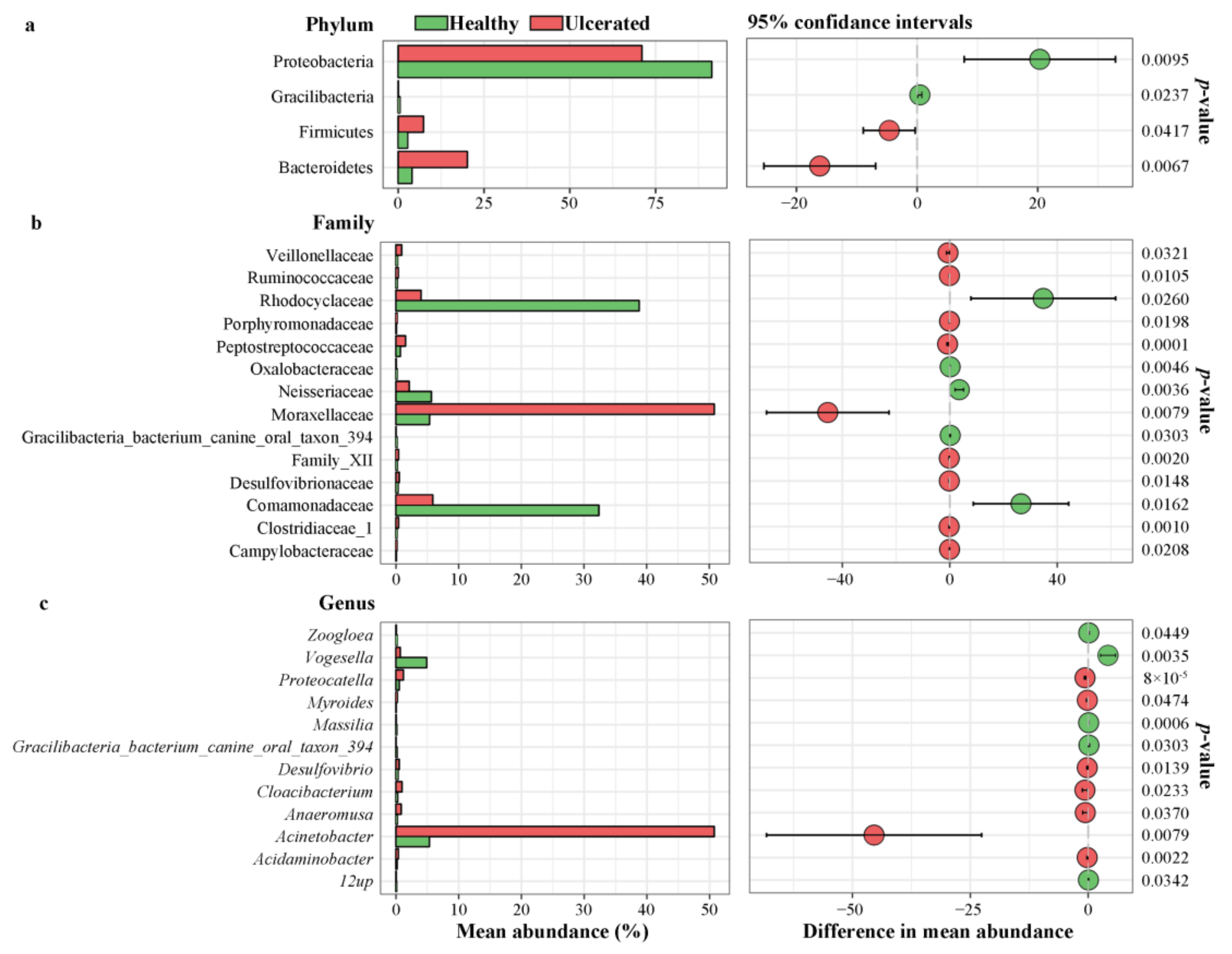
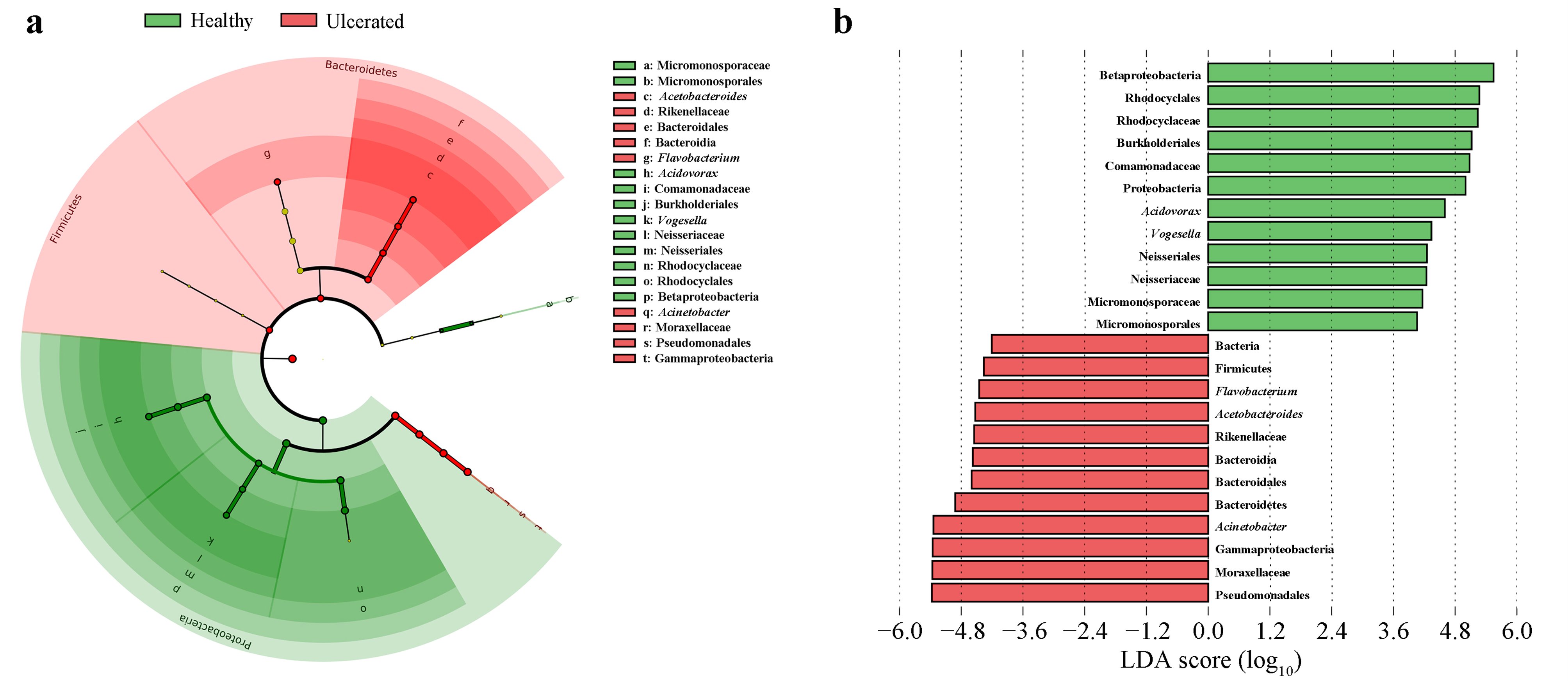
3.3. Differences in Bacterial Operational Taxonomic Units
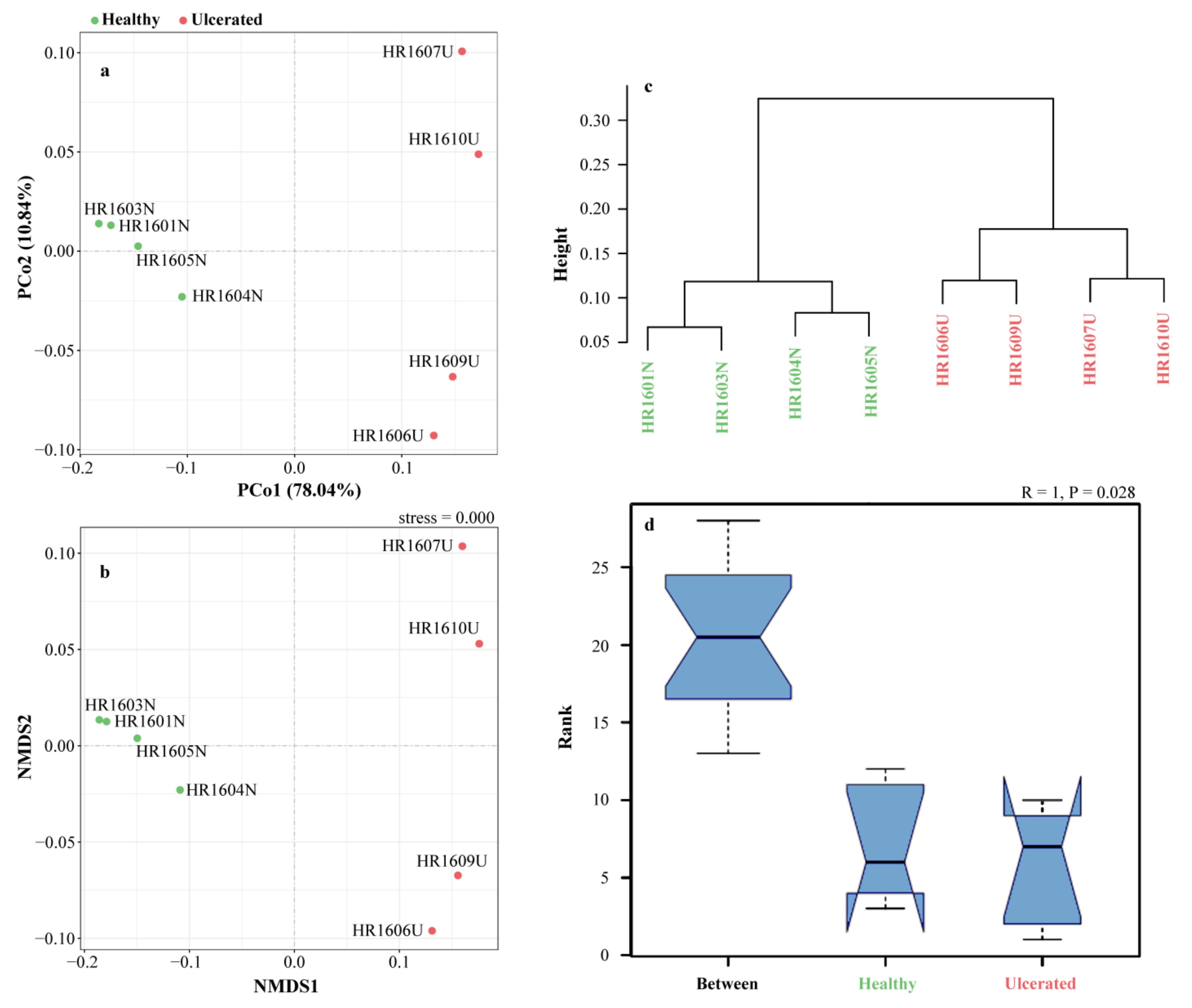
3.4. Alpha and Beta Diversity
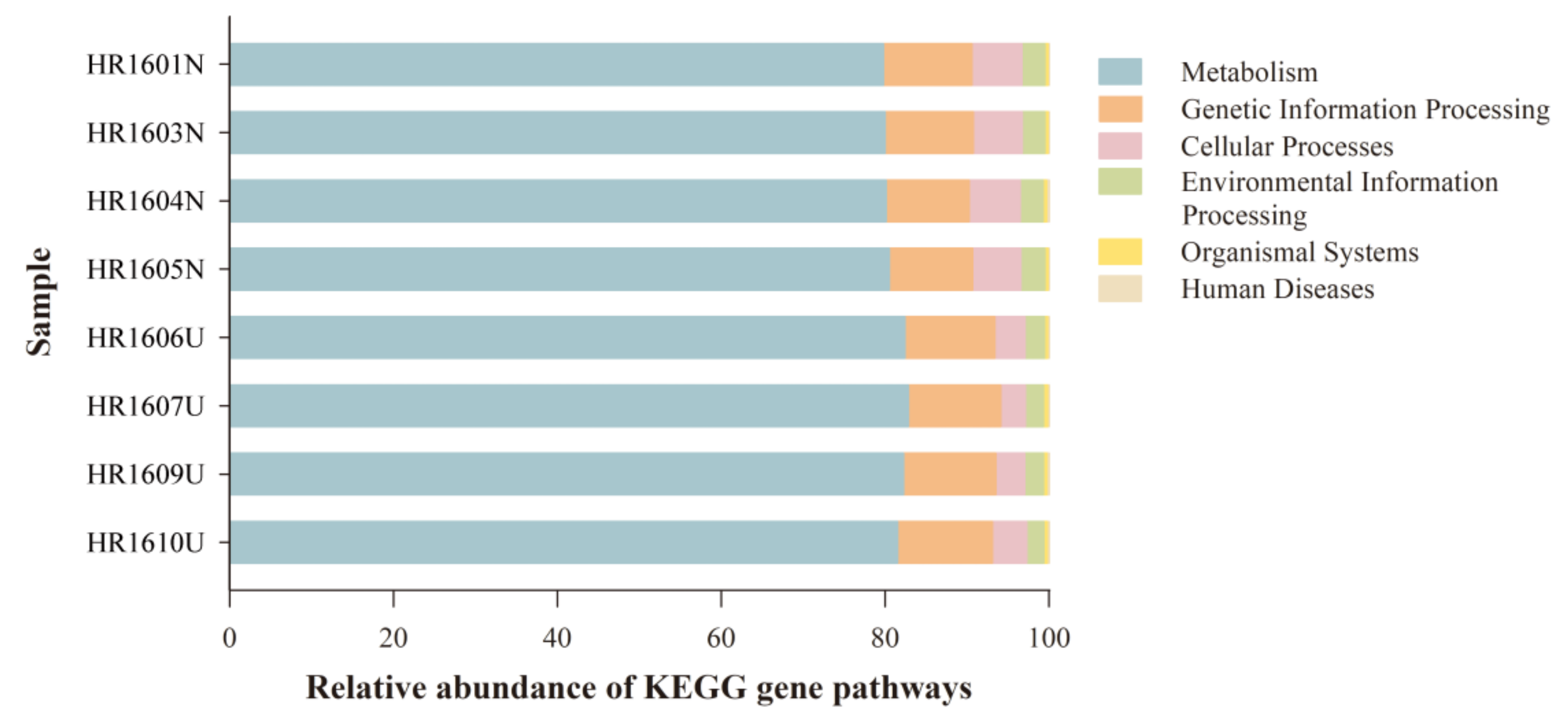
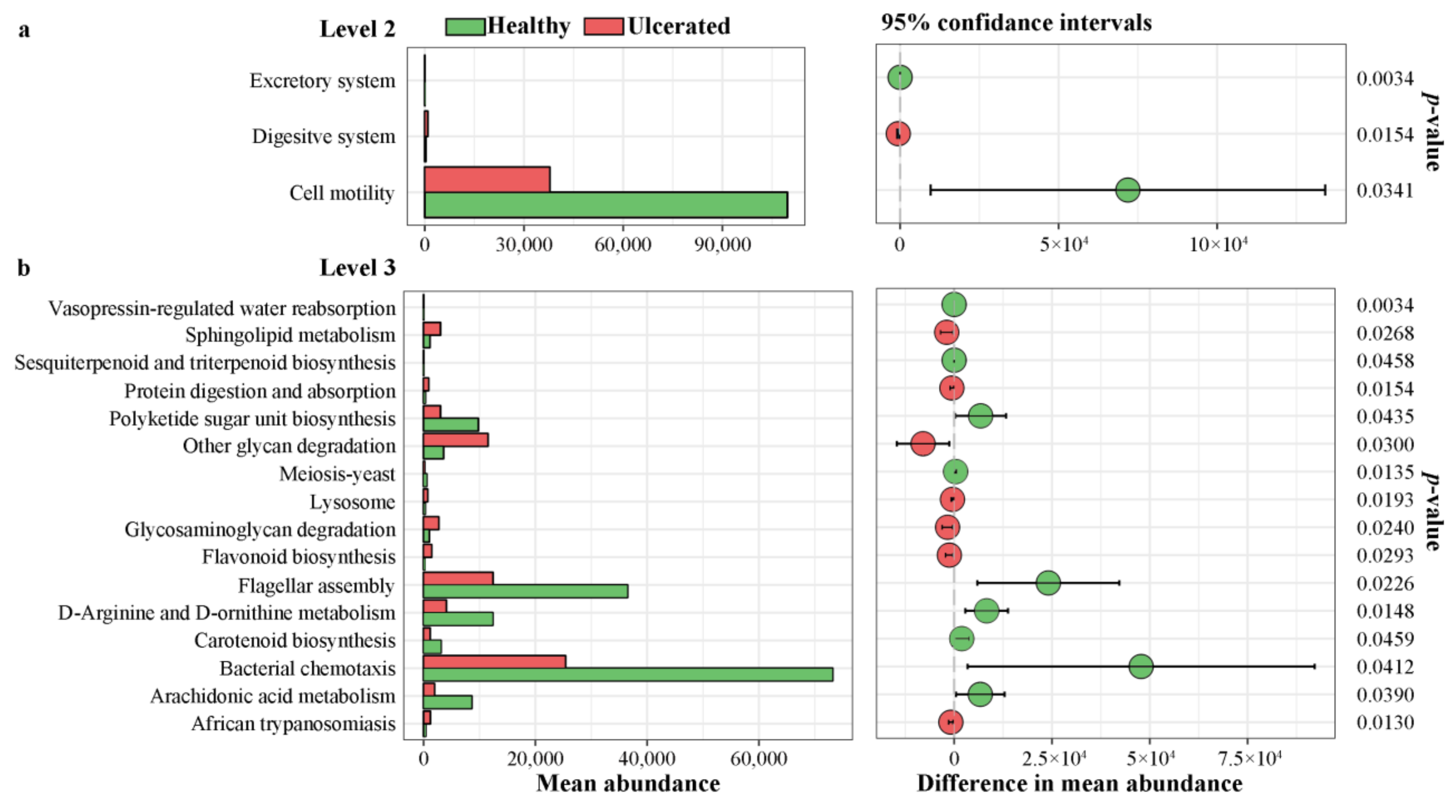
3.5. Differences in Functional Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, S.X.; Huang, X.C. Color Atlas of Practical Chinese Herbal Medicine: Animal Medicine; Guangxi Scinece & Technology Publishing House: Nanning, China, 1997. [Google Scholar]
- Yang, Z.L. Research into the development and use of China’s amphibian resources. Ecol. Econ. 2000, 11, 46–48. [Google Scholar]
- Clarke, B.T. The natural history of amphibian skin secretions, their normal functioning and potential medical applications. Biol. Rev. 2010, 72, 365–379. [Google Scholar] [CrossRef]
- Ding, G.H.; Lin, Z.H.; Fan, X.L.; Ji, X. The combined effects of food supply and larval density on survival, growth and metamorphosis of Chinese tiger frog (Hoplobatrachus rugulosa) tadpoles. Aquaculture 2015, 435, 398–402. [Google Scholar] [CrossRef]
- Pounds, J.; Masters, K. Amphibian mystery misread. Nature 2009, 462, 38–39. [Google Scholar] [CrossRef][Green Version]
- Blaustein, A.R.; Kiesecker, J.M. Complexity in conservation: Lessons from the global decline of amphibian populations. Ecol. Lett. 2002, 5, 597–608. [Google Scholar] [CrossRef]
- Poynton, S.L.; Whitaker, B.R. Protozoa and metazoa infecting amphibians. In Amphibian Medicine and Captive Husbandry; Wright, K.M., Whitaker, B.R., Eds.; Krieger Publishing Company: Malabar, FL, USA, 2001; pp. 193–221. [Google Scholar]
- Cooper, J.E.; Needham, J.R.; Griffin, J. A bacterial disease of the Darwin’s frog (Rhinoderma darwini). Lab Anim. 1978, 12, 91–93. [Google Scholar] [CrossRef][Green Version]
- Wright, K.M.; Whitaker, B.R. Amphibian Medicine and Captive Husbandry; Krieger Publishing Company: Malabar, FL, USA, 2001. [Google Scholar]
- Piotrowski, J.S.; Annis, S.L.; Longcore, J.E. Physiology of Batrachochytrium dendrobatidis, a chytrid pathogen of amphibians. Mycologia 2004, 96, 9–15. [Google Scholar] [CrossRef]
- Pennisi, E. Life and death play out on the skins of frogs. Science 2009, 326, 507–508. [Google Scholar] [CrossRef]
- Voyles, J.; Rosenblum, E.B.; Berger, L. Interactions between Batrachochytrium dendrobatidis and its amphibian hosts: A review of pathogenesis and immunity. Microbes Infect. 2011, 13, 25–32. [Google Scholar] [CrossRef]
- Woodhams, D.C.; Bletz, M.; Kueneman, J.; McKenzie, V. Managing amphibian disease with skin microbiota. Trends Microbiol. 2016, 24, 161–164. [Google Scholar] [CrossRef]
- Gray, M.J.; Miller, D.L.; Hoverman, J.T. Ecology and pathology of amphibian ranaviruses. Dis. Aquat. Org. 2009, 87, 243–266. [Google Scholar] [CrossRef]
- Pessier, A.P. An overview of amphibian skin disease. Semin. Avian Exot. Pet Med. 2002, 11, 162–174. [Google Scholar] [CrossRef]
- Campbell, C.R.; Voyles, J.; Cook, D.I.; Dinudom, A. Frog skin epithelium: Electrolyte transport and chytridiomycosis. Int. J. Biochem. Cell B 2012, 44, 431–434. [Google Scholar] [CrossRef]
- Varga, J.F.; Bui-Marinos, M.P.; Katzenback, B.A. Frog skin innate immune defences: Sensing and surviving pathogens. Front. Immunol. 2019, 9, 3128. [Google Scholar] [CrossRef]
- Harris, R.N.; James, T.Y.; Lauer, A.; Simon, M.A.; Patel, A. Amphibian pathogen Batrachochytrium dendrobatidis is inhibited by the cutaneous bacteria of amphibian species. Ecohealth 2006, 3, 53–56. [Google Scholar] [CrossRef]
- Walke, J.B.; Belden, L.K. Harnessing the microbiome to prevent fungal infections: Lessons from amphibians. PLoS Pathog. 2016, 12, e1005796. [Google Scholar] [CrossRef]
- Federici, E.; Rossi, R.; Fidati, L.; Paracucchi, R.; Scargetta, S.; Montalbani, E.; Franzetti, A.; Porta, G.L.; Fagotti, A.; Simoncelli, F.; et al. Characterization of the skin microbiota in Italian stream frogs (Rana italica) infected and uninfected by a cutaneous parasitic disease. Microbes Environ. 2015, 30, 262–269. [Google Scholar] [CrossRef]
- Campbell, L.J.; Garner, T.; Hopkins, K.; Griffiths, A.; Harrison, X.A. Outbreaks of an emerging viral disease covary with differences in the composition of the skin microbiome of a wild United Kingdom amphibian. Front. Microbiol. 2019, 10, 1245. [Google Scholar] [CrossRef]
- Harrison, X.A.; Price, S.J.; Hopkins, K.; Leung, W.T.M.; Sergeant, C.; Garner, T.W.J. Diversity-stability dynamics of the amphibian skin microbiome and susceptibility to a lethal viral pathogen. Front. Microbiol. 2019, 10, 2883. [Google Scholar] [CrossRef]
- Wake, D.B.; Vredenburg, V.T. Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc. Natl. Acad. Sci. USA 2008, 105, 11466–11473. [Google Scholar] [CrossRef]
- Fisher, M.C.; Garner, T.W.J.; Walker, S.F. Global emergence of Batrachochytrium dendrobatidis and amphibian chytridiomycosis in space, time, and host. Annu. Rev. Microbiol. 2009, 63, 291–310. [Google Scholar] [CrossRef]
- Scheele, B.C.; Pasmans, F.; Skerratt, L.F.; Berger, L.; Martel, A.; Beukema, W.; Acevedo, A.A.; Burrowes, P.A.; Carvalho, T.; Catenazzi, A.; et al. Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 2019, 363, 1459–1463. [Google Scholar] [CrossRef]
- Becker, M.H.; Harris, R.N.; Minbiole, K.; Schwantes, C.R.; Rollins-Smith, L.A.; Reinert, L.K.; Brucker, R.M.; Domangue, R.J.; Gratwicke, B. Towards a better understanding of the use of probiotics for preventing chytridiomycosis in panamanian golden frogs. Ecohealth 2011, 8, 501–506. [Google Scholar] [CrossRef]
- Becker, M.H.; Richards-Zawacki, C.L.; Gratwicke, B.; Belden, L.K. The effect of captivity on the cutaneous bacterial community of the critically endangered Panamanian golden frog (Atelopus zeteki). Biol. Conserv. 2014, 176, 199–206. [Google Scholar] [CrossRef]
- Kueneman, J.G.; Woodhams, D.C.; Harris, R.; Archer, H.M.; Knight, R.; Mckenzie, V.J. Probiotic treatment restores protection against lethal fungal infection lost during amphibian captivity. Proc. R. Soc. B 2016, 283, 20161553. [Google Scholar] [CrossRef]
- Fu, S.H. Investigation on the prevalence of bacterial infectious diseases of Rana tigerina rugulosa in Hainan Province from 2003 to 2004. Master’s Thesis, Nanjing Agricultural University, Nanjing, China, 2015. [Google Scholar]
- Armougom, F.; Raoult, D. Exploring microbial diversity using 16s rRNA high-throughput methods. J. Comput. Sci. Syst. Biol. 2009, 2, 74–92. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporas, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucle. Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Boil. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Weber, C.F. Introducing mothur: Opensource, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; The R Foundation for Statistical Computing: Vienna, Austria, 2009; Available online: http://www.R-project.org (accessed on 2 April 2020).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Wagner, H.H. Vegan: Community Ecology Package. R Package Version 2.0-10. 2013. Available online: http://sortie-admin.readyhosting.com/lme/R%20Packages/vegan.pdf (accessed on 2 April 2020).
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Huttenhower, C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef]
- Tapia-Paniagua, S.T.; Ceballos-Francisco, D.; Balebona, M.C.; María, Á.E.; Miguel, Á.M. Mucus glycosylation, immunity and bacterial microbiota associated to the skin of experimentally ulcered gilthead seabream (Sparus aurata). Fish Shellfish Immun. 2018, 75, 381–390. [Google Scholar] [CrossRef]
- Li, Z.F.; Wang, G.J.; Zhang, K.; Dong, W.B.; Yu, E.M.; Tian, J.J.; Xie, J.; Yu, D.G. Epizootic ulcerative syndrome causes cutaneous dysbacteriosis in hybrid snakehead (Channa maculate ♀ × Channa argus ♂). PeerJ 2019, 7, e6674. [Google Scholar] [CrossRef]
- Larsen, L.O. Physiology of moulting. In Physiology of the Amphibia; Moore, J.A., Lofts, B., Eds.; Academic Press: New York, NY, USA, 1976; Volume 3, pp. 53–100. [Google Scholar]
- Ohmer, M.E.B.; Cramp, R.L.; Russo, C.J.M.; White, C.R.; Franklin, C.E. Skin sloughing in susceptible and resistant amphibians regulates infection with a fungal pathogen. Sci. Rep. 2017, 7, 3529. [Google Scholar] [CrossRef]
- Sherwani, M.A.; Tufail, S.; Muzaffar, A.F.; Yusuf, N. The skin microbiome and immune system: Potential target for chemoprevention? Photodermatol. Photoimmunol. Photomed. 2018, 34, 25–34. [Google Scholar] [CrossRef]
- Chen, Y.; Fischbach, M.; Belkaid, Y. Skin microbiota—Host interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef]
- Bates, K.A.; Clare, F.C.; O’Hanlon, S.; Bosch, J.; Brookes, L.; Hopkins, K.; McLaughlin, E.J.; Daniel, O.; Garner, T.W.J.; Fisher, M.C.; et al. Amphibian chytridiomycosis outbreak dynamics are linked with host skin bacterial community structure. Nat. Commun. 2018, 9, 693. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Fraune, S.; Anton-Erxleben, F.; Augustin, R.; Franzenburg, S.; Knop, M.; Schröder, K.; Willoweit-Ohl, D.; Bosch, T.C. Bacteria-bacteria interactions within the microbiota of the ancestral metazoan Hydra contribute to fungal resistance. ISME J. 2015, 9, 1543–1556. [Google Scholar] [CrossRef]
- Sehnal, L.; Brammer-Robbins, E.; Wormington, A.M.; Blaha, L.; Bisesi, J.; Larkin, I.; Martyniuk, C.J.; Simonin, M.; Adamovsky, O. Microbiome composition and function in aquatic vertebrates: Small organisms making big impacts on aquatic animal health. Front. Microbiol. 2021, 12, 358. [Google Scholar] [CrossRef]
- Jani, A.J.; Briggs, C.J. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc. Natl. Acad. Sci. USA 2014, 111, 5049–5058. [Google Scholar] [CrossRef]
- Xu, J.X.; Xie, J.P.; Wang, Z.J. Diversity of microflora involved in the skin ulcer and death of Andrias davidianus. J. Fish. China 2021. [Google Scholar] [CrossRef]
- Kueneman, J.G.; Parfrey, L.W.; Woodhams, D.C.; Archer, H.M.; Knight, R.; McKenzie, V.J. The amphibian skinassociated microbiome across species, space and life history stages. Mol. Ecol. 2014, 23, 1238–1250. [Google Scholar] [CrossRef]
- Bletz, M.C.; Vences, M.; Sabino-Pinto, J.; Taguchi, Y.; Shimizu, N.; Nishikawa, K.; Kurabayashi, A. Cutaneous microbiota of the Japanese giant salamander (Andrias japonicus), a representative of an ancient amphibian clade. Hydrobiologia 2017, 795, 153–167. [Google Scholar] [CrossRef]
- Varela, B.J.; Lesbarrères, D.; Ibáez, R.; Green, D.M. Environmental and host effects on skin bacterial community composition in Panamanian frogs. Front. Microbiol. 2018, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Hou, X.L.; Wei, L.; Li, Y.; Qin, M.S.; Song, T.J.; Li, Y.M. Characterization of skin symbiotic bacteria of sympatric amphibians in Southeastern China. Asian Herpetol. Res. 2020, 11, 381–393. [Google Scholar]
- Mckenzie, V.J.; Bowers, R.M.; Fierer, N.; Knight, R.; Lauber, C.L. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J. 2012, 6, 588–596. [Google Scholar] [CrossRef]
- Karlsen, C.; Ottem, K.F.; Brevik, Ø.J.; Davey, M.; Sørum, H.; Winther-Larsen, H.C. The environmental and host-associated bacterial microbiota of Arctic seawater-farmed Atlantic salmon with ulcerative disorders. J. Fish. Dis. 2017, 40, 1645–1663. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef]
- Becker, M.H.; Walke, J.B.; Cikanek, S.; Savage, A.E.; Mattheus, N.; Santiago, C.N.; Minbiole, K.P.C.; Harris, R.N.; Belden, L.K.; Gratwicke, B. Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proc. R. Soc. B. 2015, 282, 20142881. [Google Scholar] [CrossRef]
- Scharschmidt, T.C.; Fischbach, M.A. What lives on our skin: Ecology, genomics and therapeutic opportunities of the skin microbiome. Drug Discov. Today Dis. Mech. 2013, 10, 83–89. [Google Scholar] [CrossRef]
- Tsoi, Y. Effect of Batrachochytrium dendrobatidis on Anura populations Batrachochytrium. IOP Conf. Ser. Earth Environ. Sci. 2021, 621, 012186. [Google Scholar] [CrossRef]
- Bierlch, K.C. Geographic Influences on the Skin Microbiome of Humpback Whales. Ph.D. Thesis, Duke University, Durham, NC, USA, 2016. [Google Scholar]
- Adegoke, A.A.; Mvuyo, T.; Okoh, A.I. Ubiquitous Acinetobacter species as beneficial commensals but gradually being emboldened with antibiotic resistance genes. J. Basic Microbiol. 2012, 52, 620–627. [Google Scholar] [CrossRef]
- Bergogne-Bérézin, E.; Friedman, H.; Bendinelli, M. Acinetobacter: Biology and Pathogenesis; Springer Science: Cham, Switzerland, 2008. [Google Scholar]
- Peleg, A.Y.; de Breij, A.; Adams, M.D.; Cerqueira, G.M.; Mocali, S.; Marco Galardini, M.; Nibbering, P.H.; Earl, A.M.; Ward, D.V.; Paterson, D.L.; et al. The success of Acinetobacter species; genetic, metabolic and virulence attributes. PLoS ONE 2012, 7, e46984. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Vuotto, C.; Donelli, G. Biofilm formation in Acinetobacter baumannii. New Microbiol. 2014, 37, 119–127. [Google Scholar] [PubMed]
- Becker, M.H.; Walke, J.B.; Murrill, L.; Woodhams, D.C.; Reinert, L.K.; Rollins-Smith, L.A.; Burzynski, E.A.; Umile, T.P.; Minbiole, K.P.C.; Belden, L.K. Phylogenetic distribution of symbiotic bacteria from Panamanian amphibians that inhibit growth of the lethal fungal pathogen Batrachochytrium dendrobatidis. Mol. Ecol. 2015, 24, 1628–1641. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gómez, O.; Kimble, S.J.A.; Briggler, J.T.; Williams, R.N. Characterization of the cutaneous bacterial communities of two giant salamander subspecies. Microbiol. Ecol. 2017, 73, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Antwis, R.E.; Preziosi, R.F.; Harrison, X.A.; Garner, T.W. Amphibian symbiotic bacteria do not show a universal ability to inhibit growth of the global panzootic lineage of Batrachochytrium dendrobatidis. Appl. Environ. Microbiol. 2015, 81, 3706–3711. [Google Scholar] [CrossRef][Green Version]
- Madison, J.D.; Berg, E.A.; Abarca, J.G.; Whitfield, S.M.; Gorbatenko, O.; Pinto, A.; Kerby, J.L. Characterization of Batrachochytrium dendrobatidis inhibiting bacteria from amphibian populations in Costa Rica. Front. Microbiol. 2017, 8, 290. [Google Scholar] [CrossRef]
- Niederle, M.V.; Bosch, J.; Ale, C.E.; Nader-Macías, M.E.; Ficoseco, C.A.; Toledo, L.F.; Valenzuela-Sánchez, A.; Soto-Azat, C.; Pasteris, S.E. Skin-associated lactic acid bacteria from North American bullfrogs as potential control agents of Batrachochytrium dendrobatidis. PLoS ONE 2019, 14, e0223020. [Google Scholar] [CrossRef]
- Kerry, R.G.; Patra, J.K.; Gouda, S.; Park, Y.; Shin, H.S.; Das, G. Benefaction of probiotics for human health: A review. J. Food Drug Anal. 2018, 26, 927–939. [Google Scholar] [CrossRef]
- Bletz, M.C.; Loudon, A.H.; Becker, M.H.; Bell, S.C.; Woodhams, D.C.; Minbiole, K.P.C.; Harris, R.N. Mitigating amphibian chytridiomycosis with bioaugmentation: Characteristics of effective probiotics and strategies for their selection and use. Eol. Lett. 2013, 16, 807–820. [Google Scholar] [CrossRef]
- Hernández-González, J.C.; Martínez-Tapia, A.; Lazcano-Hernández, G.; García-Pérez, B.E.; Castrejón-Jiménez, N.S. Bacteriocins from lactic acid bacteria. A powerful alternative as antimicrobials, probiotics, and immunomodulators in veterinary medicine. Animals 2021, 11, 979. [Google Scholar] [CrossRef]
- Hossain, M.I.; Sadekuzzaman, M.; Ha, S.D. Probiotics as potential alternative biocontrol agents in the agriculture and food industries: A review. Food Res. Int. 2017, 100, 63–73. [Google Scholar] [CrossRef]
- Harris, R.N.; Lauer, A.; Simon, M.A.; Banning, J.L.; Alford, R.A. Addition of antifungal skin bacteria to salamanders ameliorates the effects of chytridiomycosis. Dis. Aquat. Org. 2009, 83, 11–16. [Google Scholar] [CrossRef]
- Brucker, R.M.; Harris, R.N.; Schwantes, C.R.; Gallaher, T.N.; Flaherty, D.C.; Lam, B.A.; Minbiole, K.P. Amphibian chemical defense: Antifungal metabolites of the microsymbiont Janthinobacterium lividum on the salamander Plethodon cinereus. J. Chem. Ecol. 2008, 34, 1422–1429. [Google Scholar] [CrossRef]
- Jørgensen, N.O.; Brandt, K.K.; Nybroe, O.; Hansen, M. Vogesella mureinivorans sp. nov., a peptidoglycan-degrading bacterium from lake water. Int. J. Syst. Evol. Microbiol. 2010, 60, 2467–2472. [Google Scholar] [CrossRef]
- Berger, L.; Hyatt, A.D.; Speare, R.; Longcore, J.E. Life cycle stages of the amphibian chytrid Batrachochytrium dendrobatidis. Dis. Aquat. Org. 2005, 68, 51–63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Shannon | Simpson | Chao 1 | ACE | Good’s Coverage |
|---|---|---|---|---|---|
| HG | 5.26 ± 0.31 4.90–5.54 | 0.91 ± 0.02 0.89–0.93 | 1369.0 ± 178.4 1118.2–1538.3 | 1416.9 ± 160.7 1181.9–1540.4 | 0.995 ± 0.001 0.993–0.996 |
| UG | 4.76 ± 0.94 3.91–5.63 | 0.85 ± 0.09 0.76–0.94 | 1359.5 ± 114.7 1239.4–1498.1 | 1360.6 ± 111.4 1240.2–1457.3 | 0.995 ± 0.001 0.994–0.995 |
| Student’s t test | t = 1.01, p = 0.350 | t = 1.24, p = 0.261 | t = 0.09, p = 0.931 | t = 0.58, p = 0.586 | t = 0.31, p = 0.768 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.-L.; Chen, J.-M.; Chen, J.-Y.; Seah, R.W.X.; Ding, G.-H. Microbial Diversity of the Chinese Tiger Frog (Hoplobatrachus rugulosus) on Healthy versus Ulcerated Skin. Animals 2022, 12, 1241. https://doi.org/10.3390/ani12101241
Hu H-L, Chen J-M, Chen J-Y, Seah RWX, Ding G-H. Microbial Diversity of the Chinese Tiger Frog (Hoplobatrachus rugulosus) on Healthy versus Ulcerated Skin. Animals. 2022; 12(10):1241. https://doi.org/10.3390/ani12101241
Chicago/Turabian StyleHu, Hua-Li, Jia-Meng Chen, Jing-Yi Chen, Rachel Wan Xin Seah, and Guo-Hua Ding. 2022. "Microbial Diversity of the Chinese Tiger Frog (Hoplobatrachus rugulosus) on Healthy versus Ulcerated Skin" Animals 12, no. 10: 1241. https://doi.org/10.3390/ani12101241
APA StyleHu, H.-L., Chen, J.-M., Chen, J.-Y., Seah, R. W. X., & Ding, G.-H. (2022). Microbial Diversity of the Chinese Tiger Frog (Hoplobatrachus rugulosus) on Healthy versus Ulcerated Skin. Animals, 12(10), 1241. https://doi.org/10.3390/ani12101241