
GPS Coordinates for Modelling Correlated Herd Effects in Genomic Prediction Models Applied to Hanwoo Beef Cattle

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
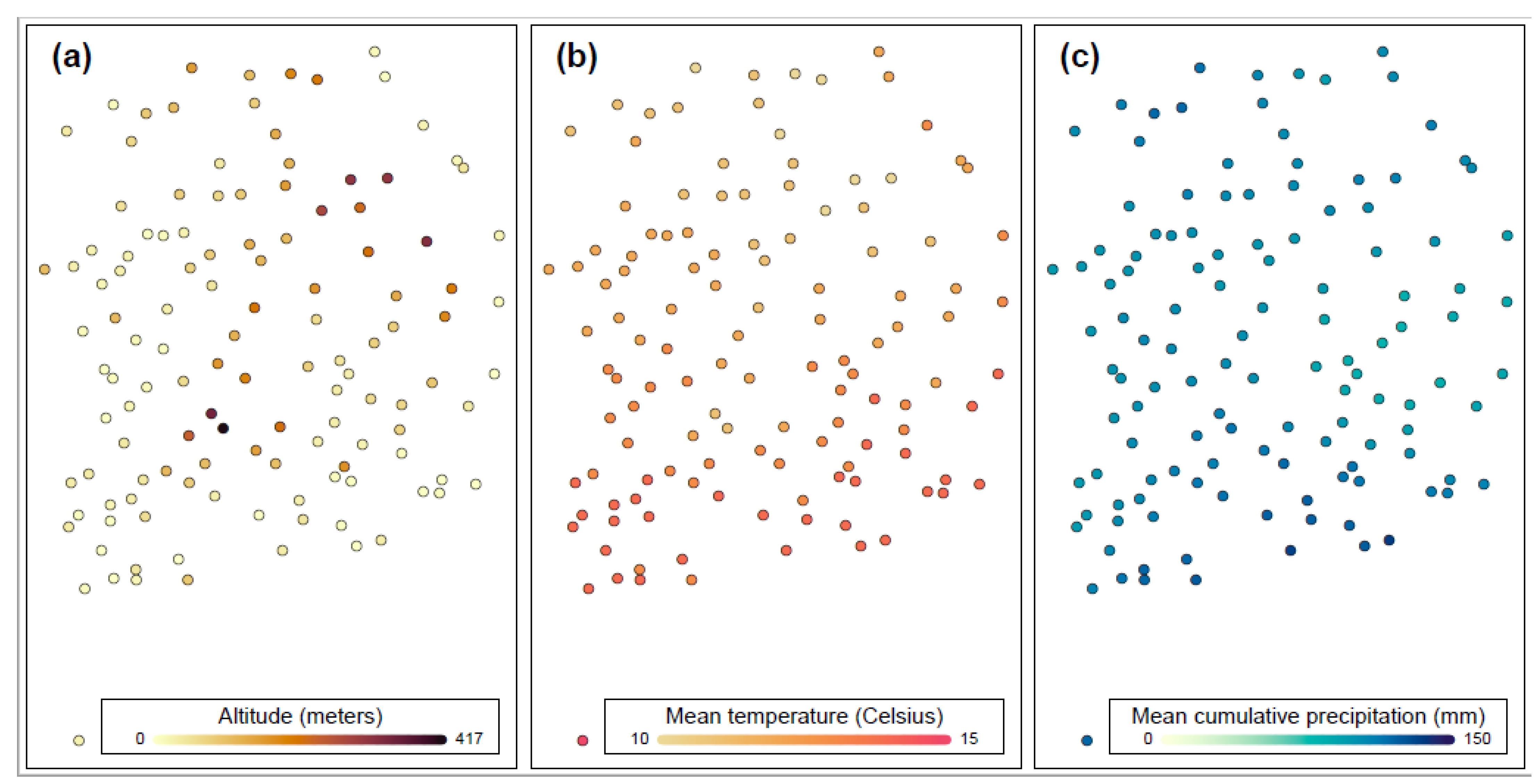
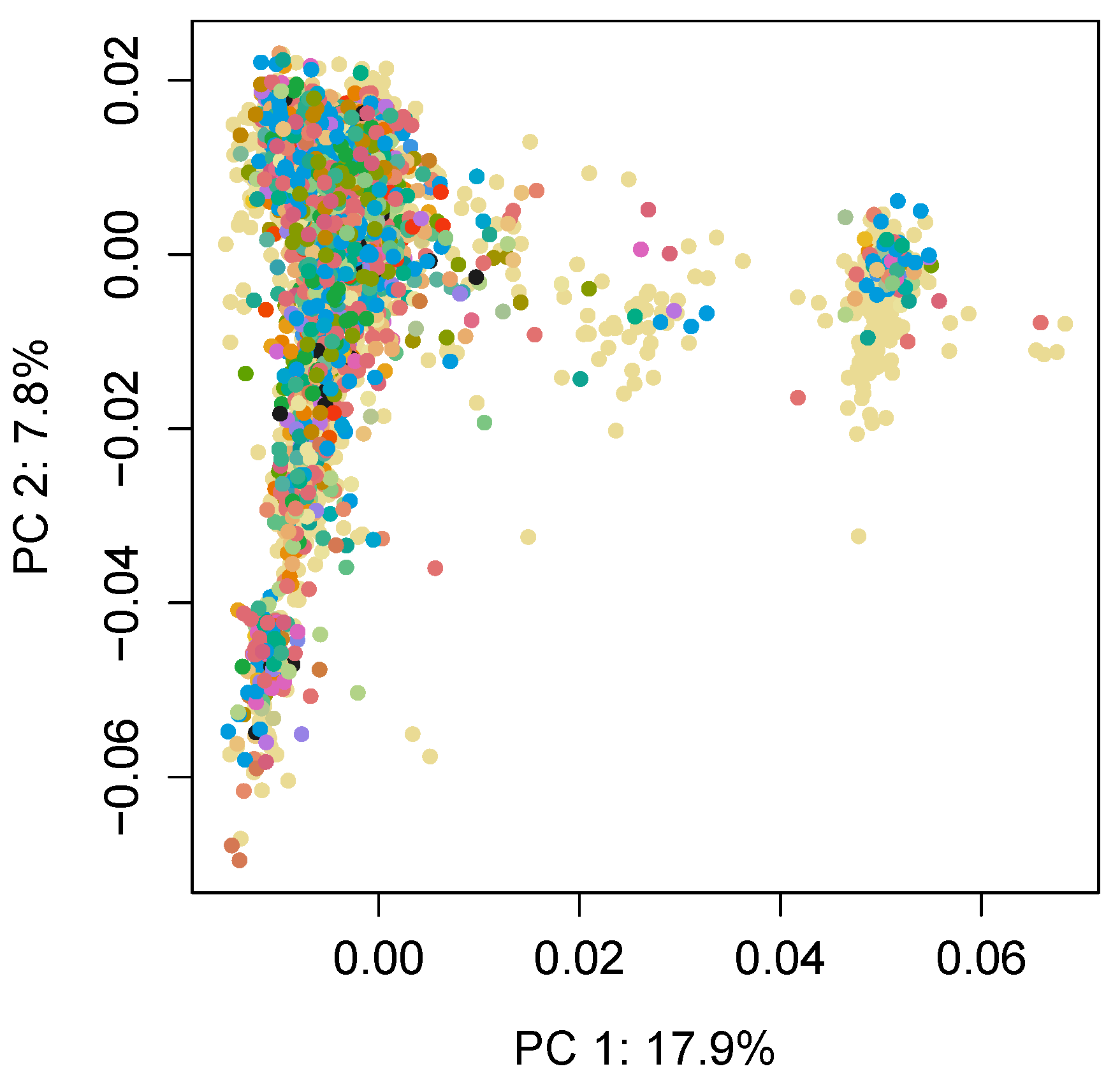
2.1. Genotypes, Phenotypes, and GPS Data
2.2. Prediction Models
2.3. Methods for Variance Components Estimation and Model Assessment
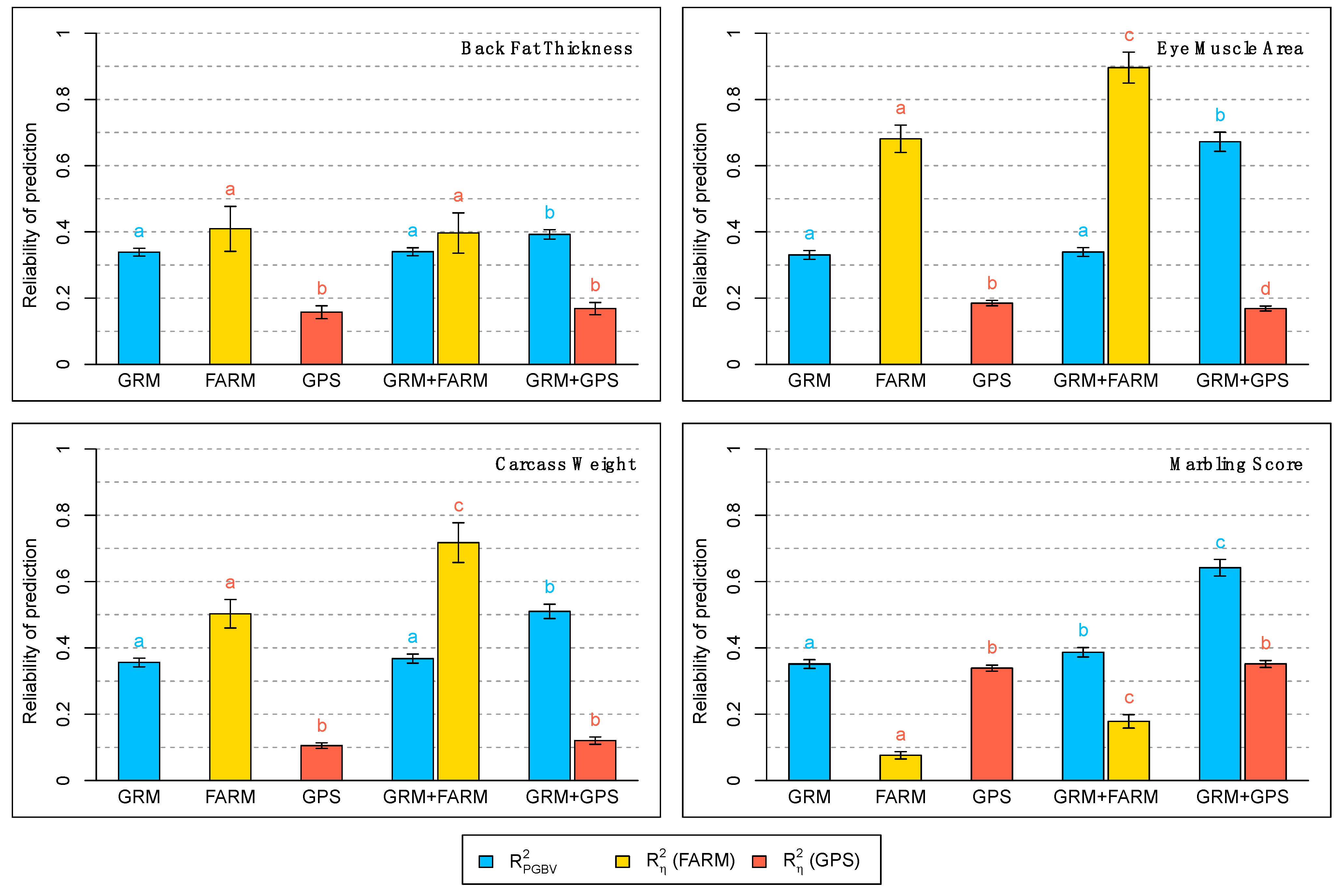
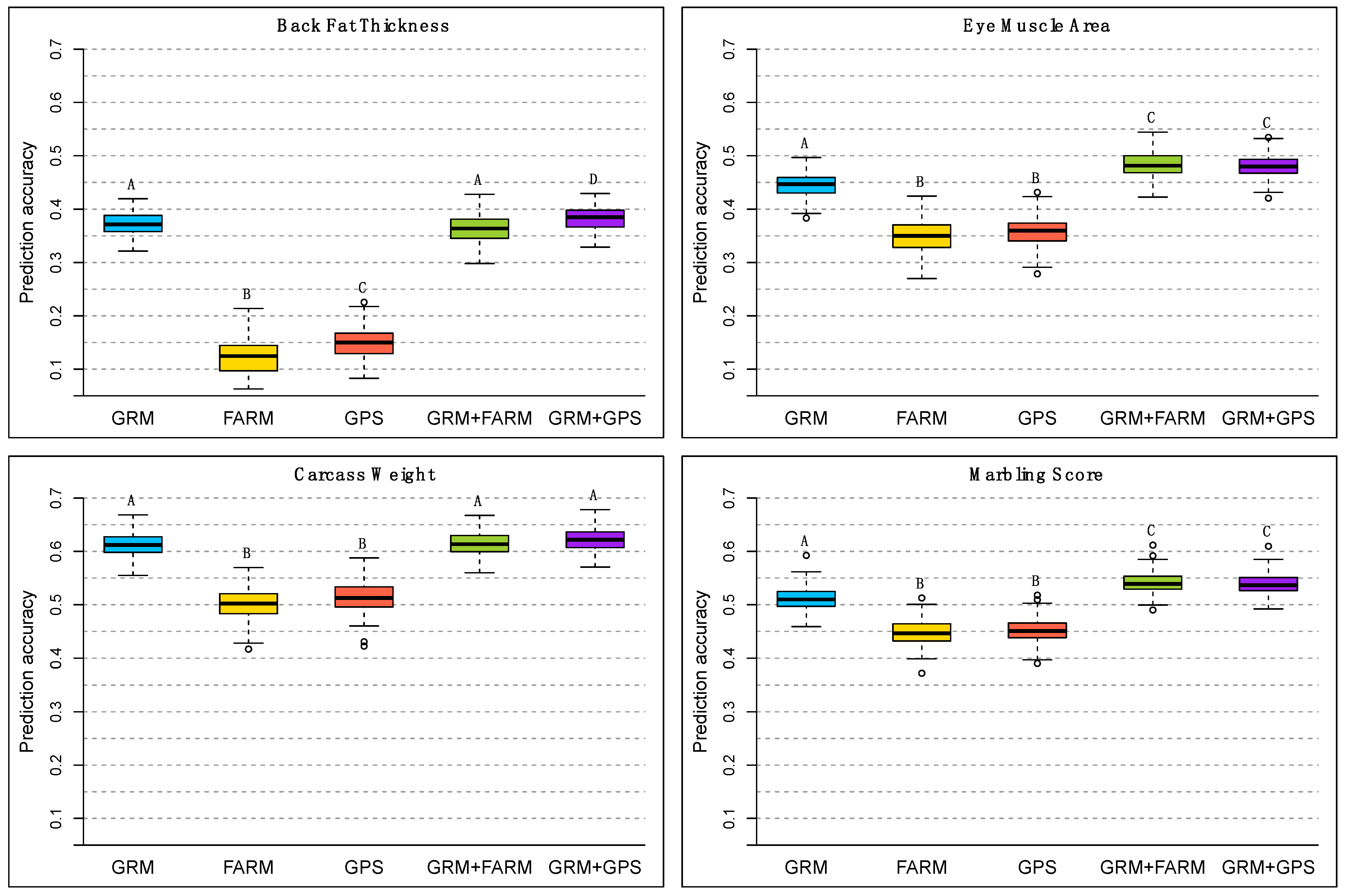
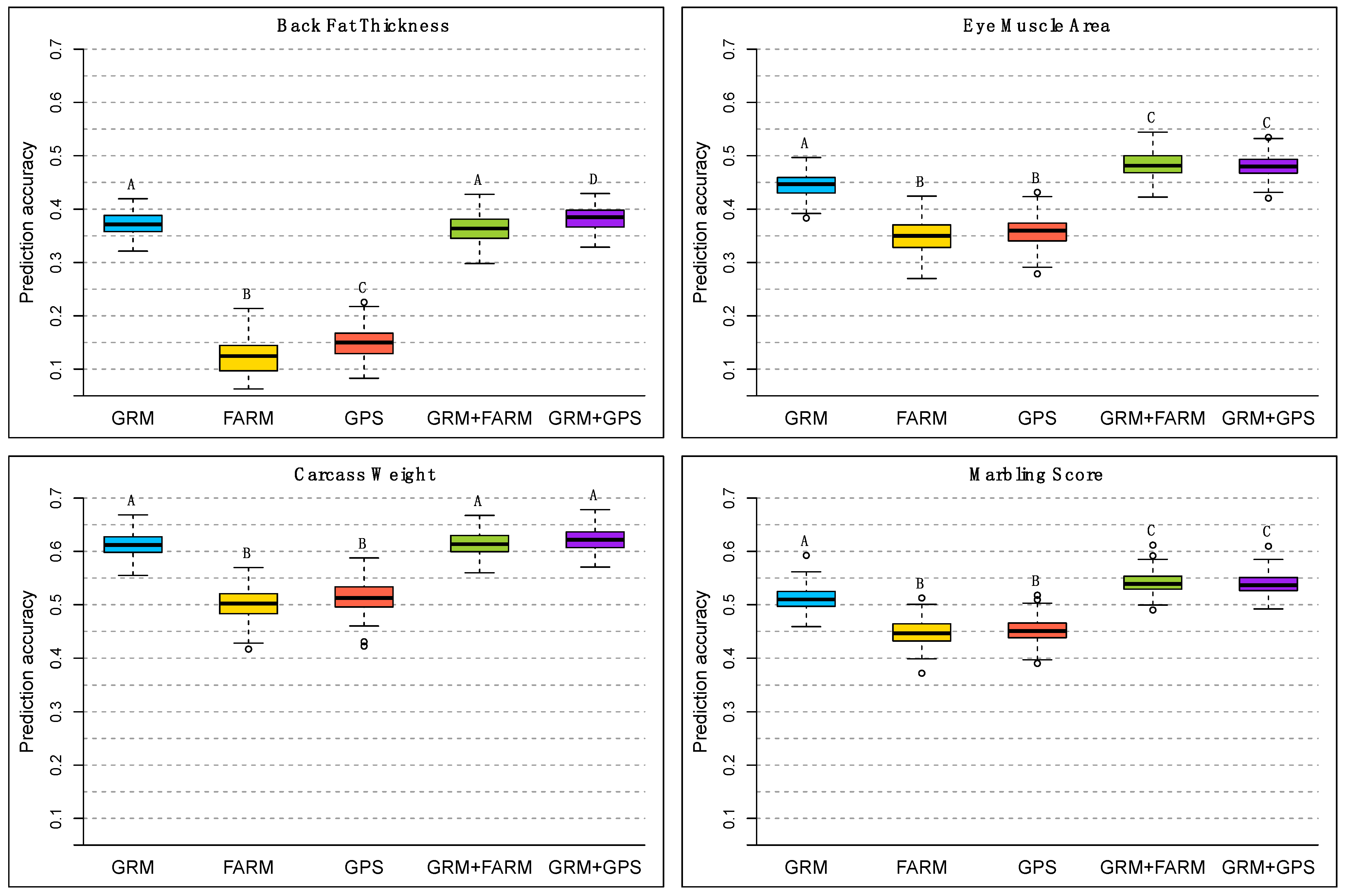
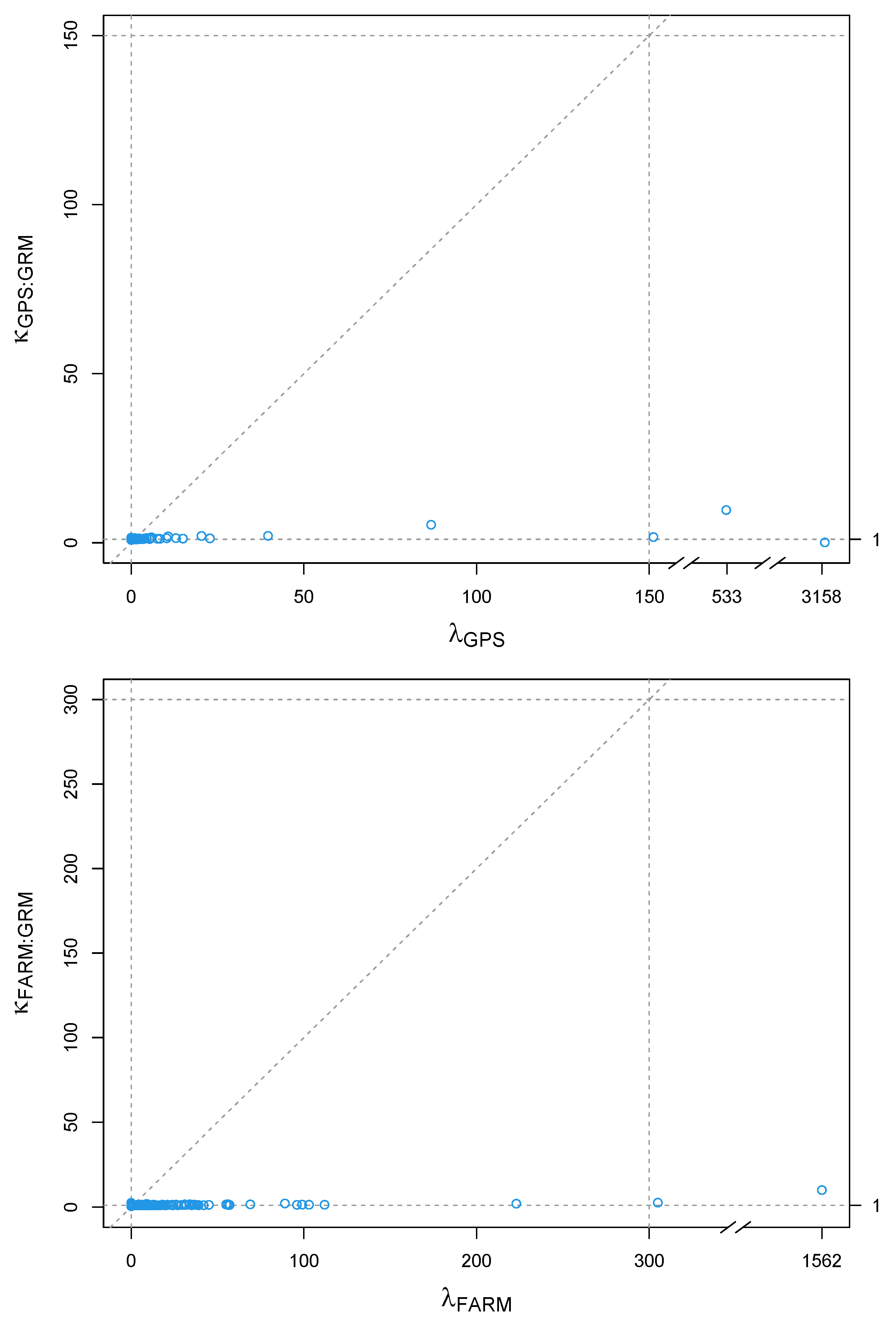
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henderson, C.R. Estimation of General, Specific and Maternal Combining Abilities in Crosses among Inbred Lines of Swine; Iowa State University: Ames, IA, USA, 1948. [Google Scholar]
- Henderson, C.R.; Kempthorne, O.; Searle, S.R.; Von Krosigk, C.M. The Estimation of Environmental and Genetic Trends from Records Subject to Culling. Biometrics 1959, 15, 192. [Google Scholar] [CrossRef]
- Wright, S. Coefficients of Inbreeding and Relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, A.; Cole, J.B.; VanRaden, P.M.; Wiggans, G.R.; Ruiz-Lopez, F.D.J.; Van Tassell, C.P. Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E3995–E4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, C.R. Sire evaluation and genetic trends. In Animal Breeding and Genetics Symposium in Honor of Dr Jay Lush; American Society in Animal Science: Champaign, IL, USA; American Society in Dairy Science Association: Champaign, IL, USA, 1973; pp. 10–41. [Google Scholar]
- Henderson, C.R. Use of Relationships Among Sires to Increase Accuracy of Sire Evaluation. J. Dairy Sci. 1975, 58, 1731–1738. [Google Scholar] [CrossRef]
- Hanocq, E.; Boichard, D.; Foulley, J. A simulation study of the effect of connectedness on genetic trend. Genet. Sel. Evol. 1996, 28, 67. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misztal, I.; Legarra, A.; Aguilar, I. Computing procedures for genetic evaluation including phenotypic, full pedigree, ad genomic information. J Dairy Sci. 2009, 92, 4648–4655. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, F.; Everett, R.; Quaas, R. Herd-Year-Season Clustering. J. Dairy Sci. 1991, 74, 629–636. [Google Scholar] [CrossRef]
- Henderson, C.R. Best Linear Unbiased Estimation and Prediction under a Selection Model. Biometrics 1975, 31, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaeffer, L.R. Contemporary Groups Are Always Random. 2009. Available online: https://animalbiosciences.uoguelph.ca/~lrs/piksLRS/ranfix.pdf (accessed on 1 July 2020).
- Schaeffer, L.R. Disconnectedness and Variance Component Estimation. Biometrics 1975, 31, 969. [Google Scholar] [CrossRef]
- Ugarte, E.; Alenda, R.; Carabaño, M. Fixed or Random Contemporary Groups in Genetic Evaluations. J. Dairy Sci. 1992, 75, 269–278. [Google Scholar] [CrossRef]
- Visscher, P.M.; Goddard, M.E. Fixed and Random Contemporary Groups. J. Dairy Sci. 1993, 76, 1444–1454. [Google Scholar] [CrossRef]
- van Bebber, J.; Reinsch, N.; Junge, W.; Kalm, E. Accounting for herd, year and season effects in genetic evaluations of dairy cattle: A review. Livest. Prod. Sci. 1997, 51, 191–203. [Google Scholar] [CrossRef]
- Tempelman, R. Addressing scope of inference for global genetic evaluation of livestock. Rev. Bras. Zootec. 2010, 39, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Tiezzi, F.; Campos, G.D.L.; Gaddis, K.P.; Maltecca, C. Genotype by environment (climate) interaction improves genomic prediction for production traits in US Holstein cattle. J. Dairy Sci. 2017, 100, 2042–2056. [Google Scholar] [CrossRef] [Green Version]
- Selle, M.L.; Steinsland, I.; Powell, O.; Hickey, J.M.; Gorjanc, G. Spatial modelling improves genetic evaluation in smallholder breeding programs. Genet. Sel. Evol. 2020, 52, 69. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, A.E.; Diggle, P.; Guttorp, P.; Fuentes, M. Handbook of Spatial Statistics; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Chiles, J.-P.; Delfiner, P. Geostatistics: Modeling Spatial Uncertainty, 2nd ed.; Wiley & Sons: Hoboken, NY, USA, 2012. [Google Scholar]
- Cressie, N. Statistics for Spatial Data, Revised ed.; Wiley: NewYork, NY, USA, 2015. [Google Scholar]
- Rue, H.; Held, L. Gaussian Markov Random Fields: Theory and Applications, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2005. [Google Scholar]
- Matérn, B. Spatial Variation, 2nd ed.; Lecture Notes in Statistics; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high-resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Fick, S.E.; Hijmans, R.J. Worldclim 2: New 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Livestock Products Grading Guideline; Minister for Food, Agriculture, Forestry and Fisheries: Seoul, Korean, 2011.
- Park, B.; Choi, T.; Kim, S.; Oh, S.-H. National Genetic Evaluation (System) of Hanwoo (Korean Native Cattle). Asian-Australas. J. Anim. Sci. 2013, 26, 151–156. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- Hijmans, R.J.; Williams. geosphere: Spherical Trigonometry. R Package Version 1.5-10. 2019. Available online: https://CRAN.R-project.org/package=geosphere (accessed on 1 July 2021).
- Patterson, H.D.; Thompson, R. Recovery of Inter-Block Information when Block Sizes are Unequal. Biometrika 1971, 58, 545–554. [Google Scholar] [CrossRef]
- Tukey, J.W. Comparing Individual Means in the Analysis of Variance. Biometrics 1949, 5, 99. [Google Scholar] [CrossRef]
- Fisher, R.A. Frequency Distribution of the Values of the Correlation Coefficient in Samples from an Indefinitely Large Population. Biometrika 1915, 10, 507. [Google Scholar] [CrossRef]
- Cuyabano, B.C.D.; Sørensen, A.C.; Sørensen, P. Understanding the potential bias of variance components estimators when using genomic models. Genet. Sel. Evol. 2018, 50, 41. [Google Scholar] [CrossRef] [Green Version]
- Rendel, J.M.; Robertson, A. Estimation of genetic gain in milk yield by selection in a closed herd of dairy cattle. J. Genet. 1950, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E. Genomic selection: Prediction of accuracy and maximization of long-term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef]
- Bijma, P. Accuracies of estimated breeding values from ordinary genetic evaluations do not reflect the correlation between true and estimated breeding values in selected populations. J. Anim. Breed. Genet. 2012, 129, 345–358. [Google Scholar] [CrossRef]
- Lozano-Jaramillo, M.; Alemu, S.W.; Dessie, T.; Komen, H.; Bastiaansen, J.W.M. Using phenotypic distribution models to predict livestock performance. Sci. Rep. 2019, 9, 15371. [Google Scholar] [CrossRef] [Green Version]
- Jarquín, D.; Da Silva, C.L.; Gaynor, R.; Poland, J.; Fritz, A.; Howard, R.; Battenfield, S.; Crossa, J. Increasing Genomic-Enabled Prediction Accuracy by Modeling Genotype × Environment Interactions in Kansas Wheat. Plant Genome 2017, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gillberg, J.; Marttinen, P.; Mamitsuka, H.; Kaski, S. Modelling G × E with historical weather information improves genomic prediction in new environments. Bioinformatics 2019, 35, 4045–4052. [Google Scholar] [CrossRef] [Green Version]
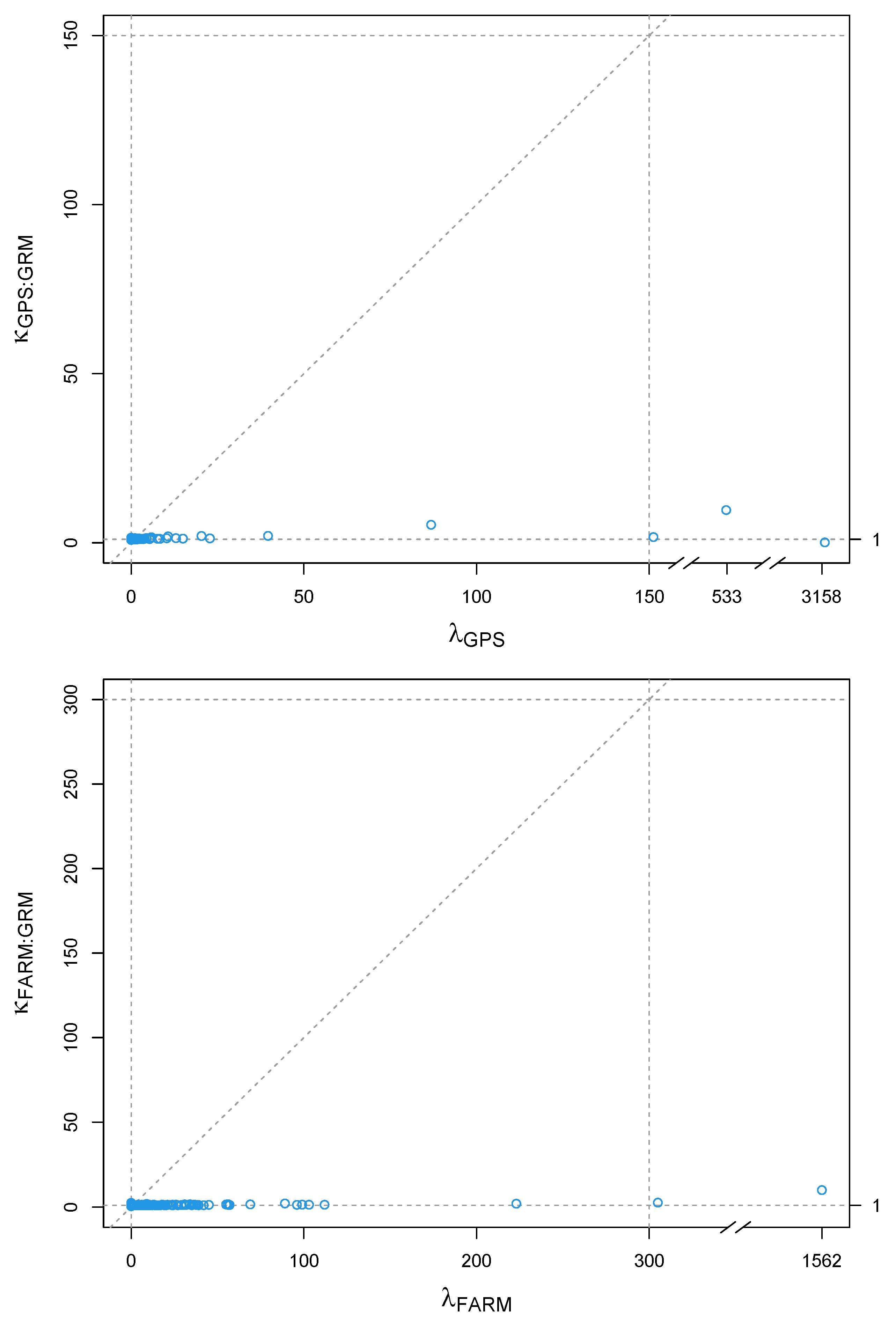

{kind=link}
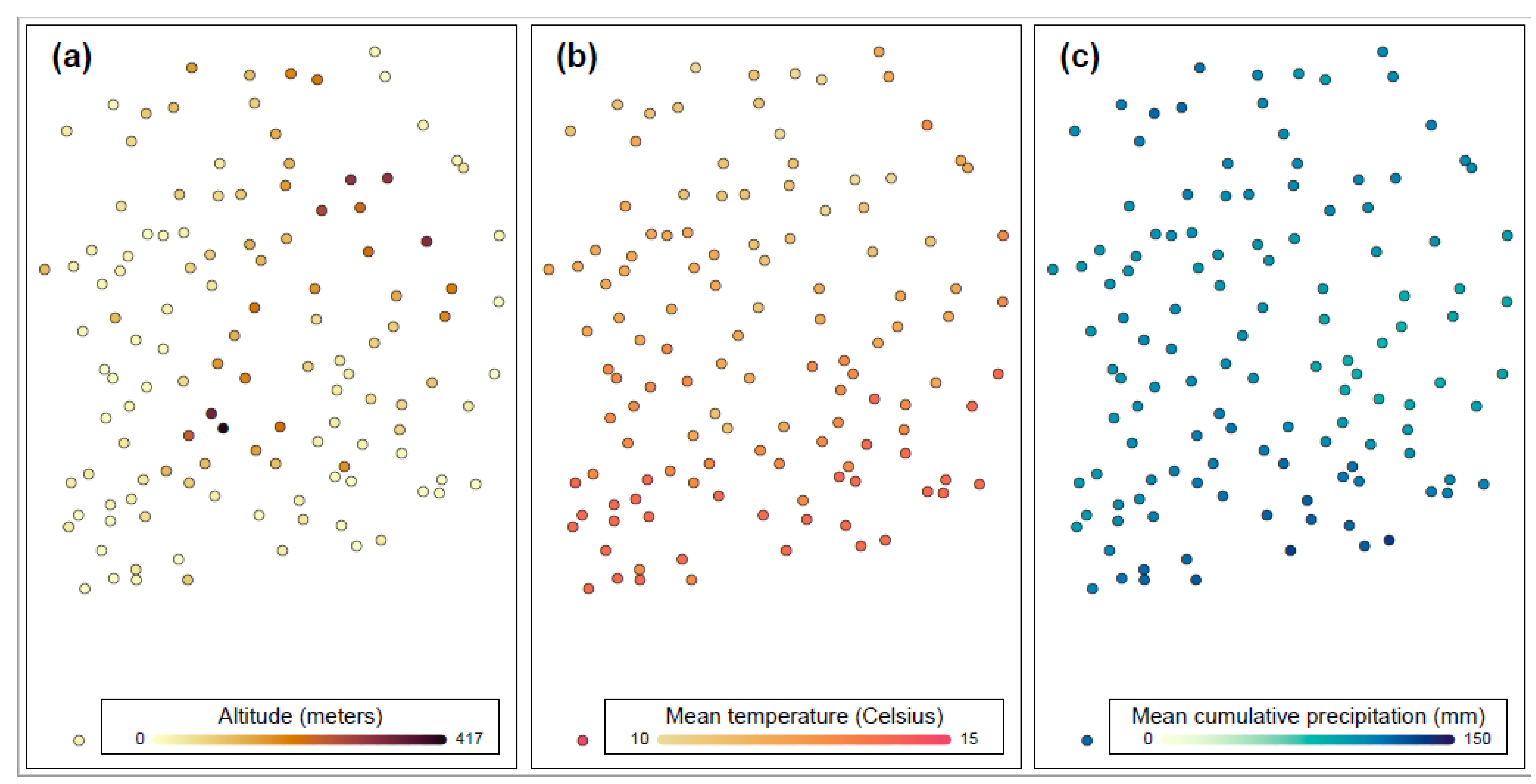
{kind=link}
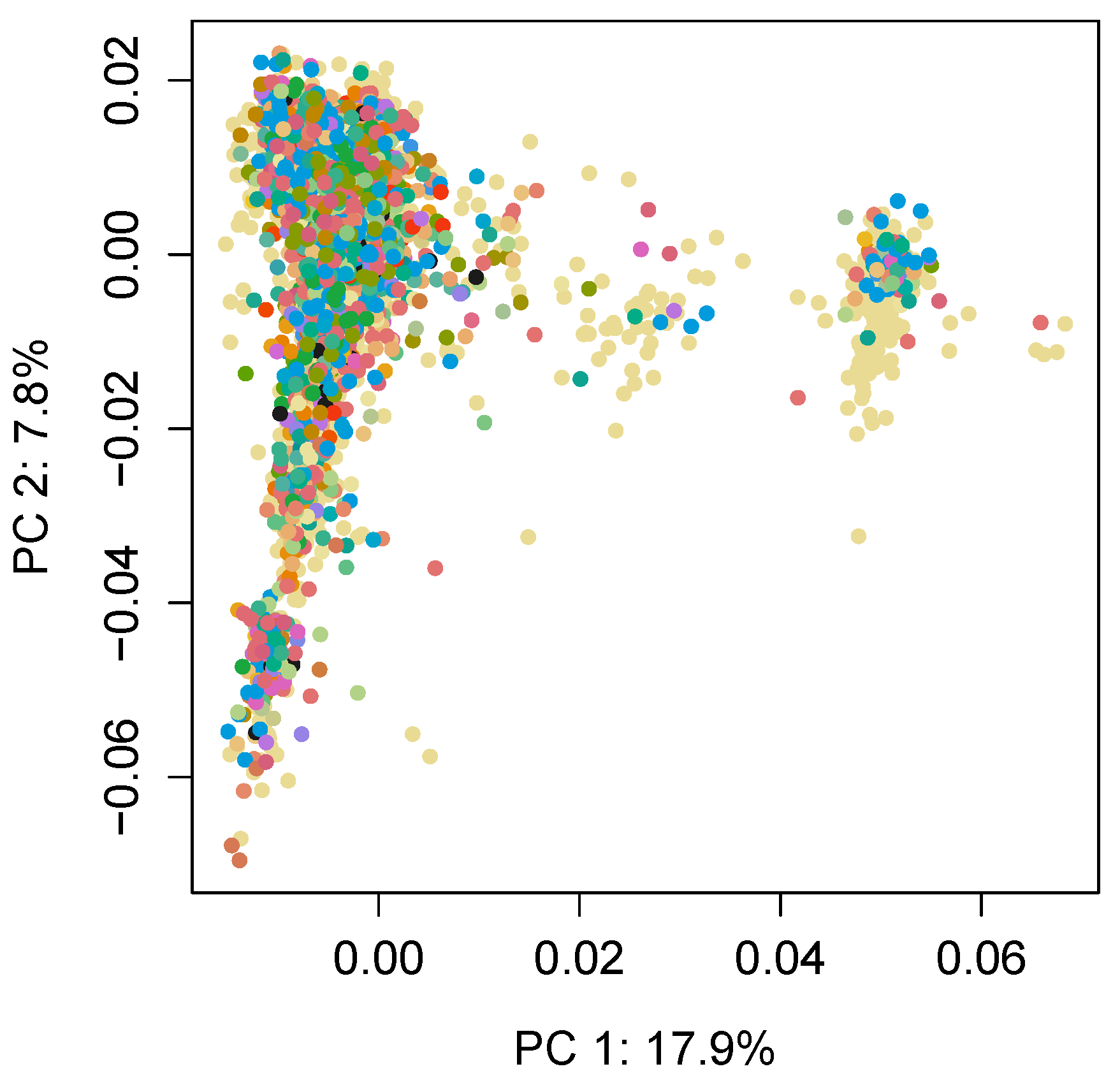
{kind=link}
{kind=link}
{kind=link}
| Sample Size | Number of Farms |
|---|---|
| <5 | 41 |
| 6–10 | 34 |
| 11–20 | 14 |
| 21–50 | 22 |
| 51–100 | 8 |
| 101–350 | 4 |
| 1562 | 1 |
| Trait | Sex | Age (Months) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 | 33 | 34 | 35 | All | ||
| Backfat Thickness (mm) | Bull | 12.9 ±4.4 | 12.8 ±2.7 | 13.9 ±5.0 | 12.1 ±3.7 | 15.1 ±6.2 | 14.0 ±5.1 | 14.2 ±4.2 | 13.1 ±4.8 | 12.6 ±3.8 | 15.3 ±5.7 | 14.2 ±5.7 | 13.9 ±5.0 |
| Steer | 12.7 ±5.5 | 13.2 ±5.4 | 13.3 ±4.8 | 14.0 ±5.0 | 14.0 ±4.8 | 14.6 ±5.1 | 14.4 ±5.1 | 15.0 ±5.0 | 14.5 ±5.1 | 14.1 ±6.0 | 14.9 ±6.2 | 14.4 ±5.1 | |
| All | 12.8 ±5.0 | 13.1 ±4.9 | 13.4 ±4.8 | 13.7 ±4.9 | 14.1 ±4.8 | 14.6 ±5.1 | 14.4 ±5.0 | 14.9 ±5.0 | 14.3 ±5.0 | 14.5 ±5.9 | 14.7 ±6.0 | 14.3 ±5.1 | |
| Eye Muscle Area (cm2) | Bull | 81.7 ±16.7 | 84.1 ±12.1 | 89.7 ±15.4 | 85.5 ±12.4 | 89.2 ±8.5 | 84.1 ±10.8 | 86.5 ±8.8 | 86.7 ±13.7 | 87.6 ±12.0 | 90.7 ±10.9 | 90.5 ±14.6 | 87.8 ±12.2 |
| Steer | 87.6 ±14.1 | 92.7 ±16.7 | 95.2 ±11.7 | 97.8 ±12.6 | 96.4 ±11.6 | 98.3 ±11.6 | 98.7 ±11.2 | 97.5 ±11.4 | 96.7 ±11.2 | 96.8 ±11.6 | 99.0 ±12.8 | 97.6 ±11.7 | |
| All | 85.5 ±15.0 | 90.9 ±16.1 | 93.8 ±12.9 | 96.1 ±13.3 | 96.0 ±11.5 | 97.9 ±11.8 | 98.2 ±11.4 | 96.9 ±11.9 | 95.6 ±11.7 | 94.7 ±11.7 | 96.0 ±14.0 | 96.7 ±12.1 | |
| Carcass Weight (kg) | Bull | 319.9 ±76.8 | 339.9 ±44.4 | 344.9 ±52.1 | 344.1 ±44.4 | 362.0 ±44.5 | 338.7 ±47.1 | 350.8 ±25.6 | 355.5 ±47.9 | 360.2 ±44.6 | 379.9 ±48.2 | 367.6 ±53.0 | 356.2 ±48.3 |
| Steer | 406.9 ±50.7 | 418.4 ±57.4 | 430.2 ±48.7 | 444.5 ±47.5 | 445.4 ±46.9 | 450.2 ±46.1 | 455.4 ±45.9 | 456.7 ±49.8 | 452.9 ±48.5 | 448.4 ±58.1 | 465.2 ±59.4 | 450.1 ±48.2 | |
| All | 375.9 ±73.4 | 402.4 ±63.2 | 408.2 ±62.0 | 430.9 ±58.2 | 440.9 ±50.4 | 447.1 ±49.7 | 450.8 ±50.0 | 450.3 ±55.4 | 441.2 ±57.0 | 425.4 ±63.7 | 430.5 ±73.8 | 442.0 ±54.9 | |
| Marbling Score (9 levels) | Bull | 4.6 ±2.1 | 4.5 ±2.2 | 5.0 ±2.2 | 4.7 ±1.9 | 5.3 ±1.6 | 5.2 ±2.2 | 5.1 ±2.2 | 4.1 ±1.9 | 4.4 ±1.9 | 5.5 ±1.8 | 4.9 ±2.0 | 4.9 ±2.0 |
| Steer | 5.1 ±1.8 | 6.4 ±2.1 | 6.4 ±1.8 | 6.8 ±1.6 | 6.5 ±1.7 | 6.7 ±1.6 | 6.6 ±1.6 | 6.5 ±1.6 | 6.4 ±1.6 | 6.6 ±1.7 | 6.5 ±1.7 | 6.6 ±1.7 | |
| All | 4.9 ±1.9 | 6.0 ±2.2 | 6.1 ±2.0 | 6.5 ±1.8 | 6.4 ±1.8 | 6.7 ±1.6 | 6.6 ±1.7 | 6.4 ±1.8 | 6.1 ±1.8 | 6.2 ±1.8 | 5.9 ±2.0 | 6.4 ±1.8 | |
| N | Bull | 10 | 10 | 34 | 41 | 44 | 27 | 39 | 30 | 31 | 54 | 38 | 358 |
| Steer | 18 | 39 | 98 | 261 | 775 | 925 | 855 | 448 | 215 | 107 | 69 | 3810 | |
| All | 28 | 49 | 132 | 302 | 819 | 952 | 894 | 478 | 246 | 161 | 107 | 4168 | |
| Model | Equation |
| GRM | |
| FARM | |
| GPS | |
| GRM + FARM | |
| GRM + GPS |
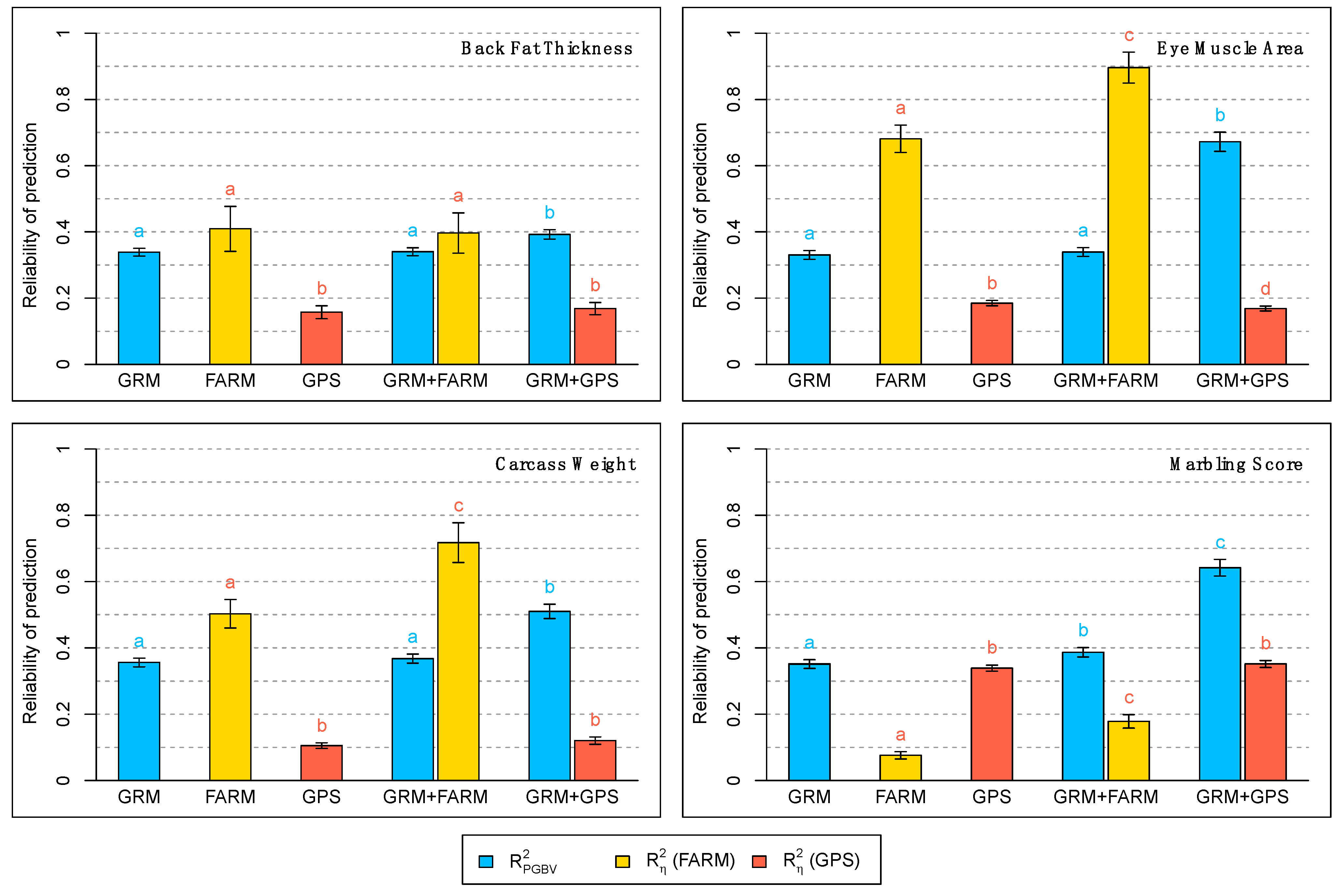
| Trait | Model | ||||||
|---|---|---|---|---|---|---|---|
| Backfat Thickness | GRM | 0.35 a,† | - | 0.34 a | - | 0.34 a | - |
| FARM | - | 0.03 a,† | - | 0.10 a | - | 0.32 a | |
| GPS | - | 0.14 b,† | - | 0.14 b | - | 0.13 b | |
| GRM + FARM | 0.35 a,† | 0.03 a,† | 0.34 a | 0.10 a | 0.34 a | 0.32 a | |
| GRM + GPS | 0.30 a,† | 0.15 b,† | 0.34 a | 0.15 b | 0.39 b | 0.15 b | |
| Eye Muscle Area | GRM | 0.35 a,† | - | 0.34 a | - | 0.33 a | - |
| FARM | - | 0.09 a,† | - | 0.24 a | - | 0.66 a | |
| GPS | - | 0.53 b,† | - | 0.31 b | - | 0.18 b | |
| GRM + FARM | 0.34 a,† | 0.09 a,† | 0.34 a | 0.28 c | 0.34 a | 0.87 c | |
| GRM + GPS | 0.17 b,† | 0.53 b,† | 0.34 a | 0.30 b | 0.66 b | 0.17 d | |
| Carcass Weight | GRM | 0.41 a,† | - | 0.38 a | - | 0.35 a | - |
| FARM | - | 0.06 a,† | - | 0.17 a | - | 0.47 a | |
| GPS | - | 0.40 b,† | - | 0.20 b | - | 0.10 b | |
| GRM + FARM | 0.39 a,† | 0.05 c,† | 0.38 a | 0.17 a | 0.36 a | 0.67 c | |
| GRM + GPS | 0.29 b,† | 0.30 d,† | 0.38 a | 0.18 a,b | 0.50 b | 0.11 b | |
| Marbling Score | GRM | 0.40 a,† | - | 0.37 a | - | 0.35 a | - |
| FARM | - | 0.12 a,† | - | 0.09 a | - | 0.07 a | |
| GPS | - | 0.47 b,† | - | 0.40 b | - | 0.34 b | |
| GRM + FARM | 0.36 a,† | 0.10 c,† | 0.37 a | 0.13 c | 0.38 b | 0.16 c | |
| GRM + GPS | 0.22 b,† | 0.44 b,† | 0.37 a | 0.39 b | 0.63 c | 0.35 b |
| Trait | Models Compared | corp | |||
|---|---|---|---|---|---|
| Model 1 | Model 2 | ||||
| Backfat Thickness | GRM | GRM + FARM | 0.98 (0.002) | 0.98 (0.002) | 0.17 (0.018) |
| GRM | GRM + GPS | 0.99 (0.001) | 0.99 (0.001) | 0.07 (0.007) | |
| Eye Muscle Area | GRM | GRM + FARM | 0.97 (0.003) | 0.97 (0.003) | 1.54 (0.167) |
| GRM | GRM + GPS | 0.99 (0.001) | 0.99 (0.001) | 0.54 (0.065) | |
| Carcass Weight | GRM | GRM + FARM | 0.99 (0.001) | 0.98 (0.002) | 14.06 (1.438) |
| GRM | GRM + GPS | 1.00 (0.000) | 1.00 (0.001) | 3.70 (0.479) | |
| Marbling Score | GRM | GRM + FARM | 0.98 (0.002) | 0.98 (0.003) | 0.02 (0.002) |
| GRM | GRM + GPS | 0.99 (0.001) | 0.99 (0.001) | 0.01 (0.001) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuyabano, B.C.D.; Rovere, G.; Lim, D.; Kim, T.H.; Lee, H.K.; Lee, S.H.; Gondro, C. GPS Coordinates for Modelling Correlated Herd Effects in Genomic Prediction Models Applied to Hanwoo Beef Cattle. Animals 2021, 11, 2050. https://doi.org/10.3390/ani11072050
Cuyabano BCD, Rovere G, Lim D, Kim TH, Lee HK, Lee SH, Gondro C. GPS Coordinates for Modelling Correlated Herd Effects in Genomic Prediction Models Applied to Hanwoo Beef Cattle. Animals. 2021; 11(7):2050. https://doi.org/10.3390/ani11072050
Chicago/Turabian StyleCuyabano, Beatriz Castro Dias, Gabriel Rovere, Dajeong Lim, Tae Hun Kim, Hak Kyo Lee, Seung Hwan Lee, and Cedric Gondro. 2021. "GPS Coordinates for Modelling Correlated Herd Effects in Genomic Prediction Models Applied to Hanwoo Beef Cattle" Animals 11, no. 7: 2050. https://doi.org/10.3390/ani11072050
APA StyleCuyabano, B. C. D., Rovere, G., Lim, D., Kim, T. H., Lee, H. K., Lee, S. H., & Gondro, C. (2021). GPS Coordinates for Modelling Correlated Herd Effects in Genomic Prediction Models Applied to Hanwoo Beef Cattle. Animals, 11(7), 2050. https://doi.org/10.3390/ani11072050