
Genome of the Southern Giant Petrel Assembled Using Third-Generation DNA Sequencing and Linked Reads Reveals Evolutionary Traits of Southern Avian

 ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Library Construction and Sequencing
2.3. Genome Assembly
2.4. Gene Prediction and Annotation
2.5. Comparative Genomics
2.6. Population History
2.7. Gene Family Identification and Phylogenetic Analysis
2.8. Simple Sequence Repeat Identification
3. Results
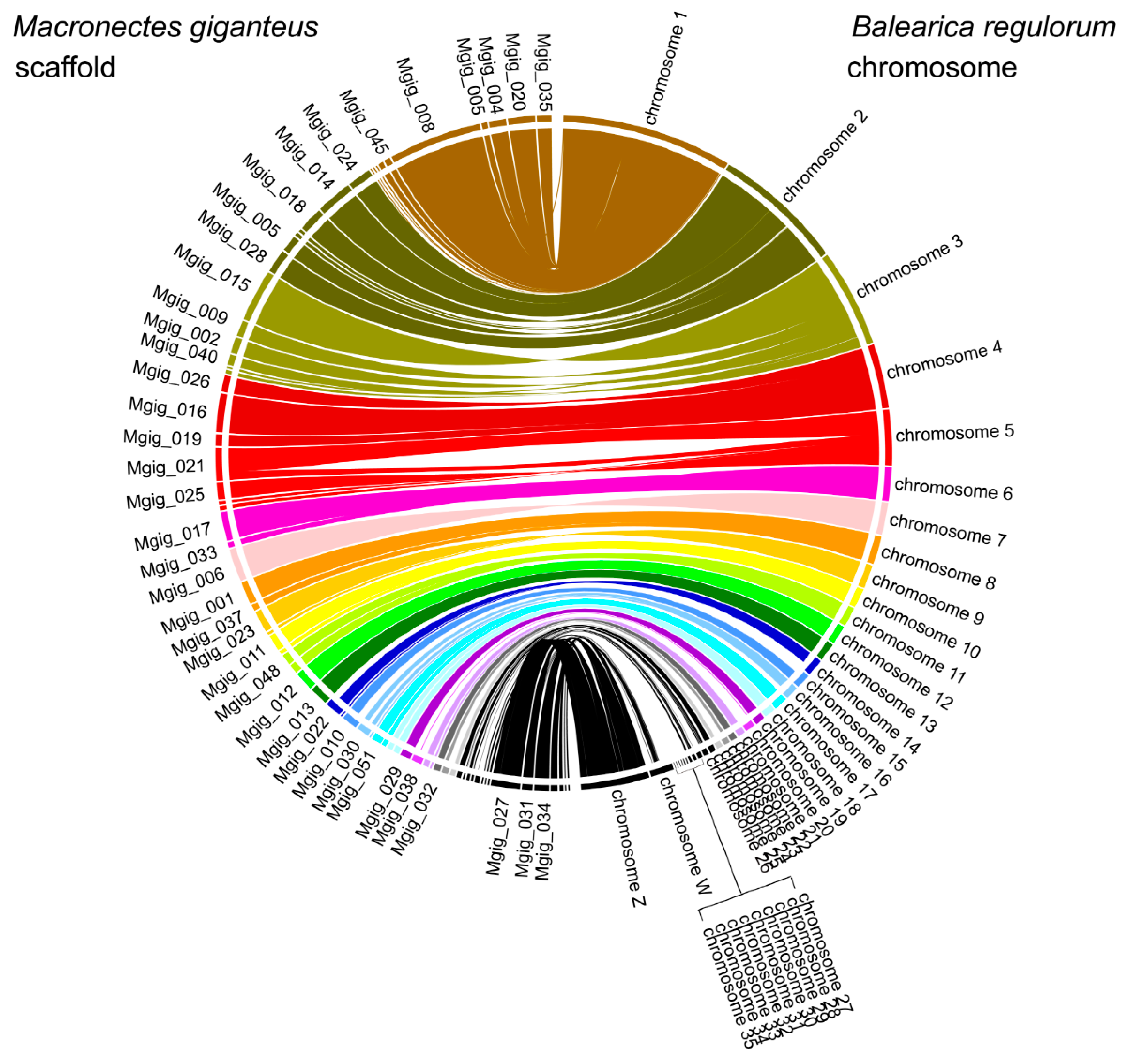
3.1. Genome Assembly
3.2. Gene Prediction and Annotation
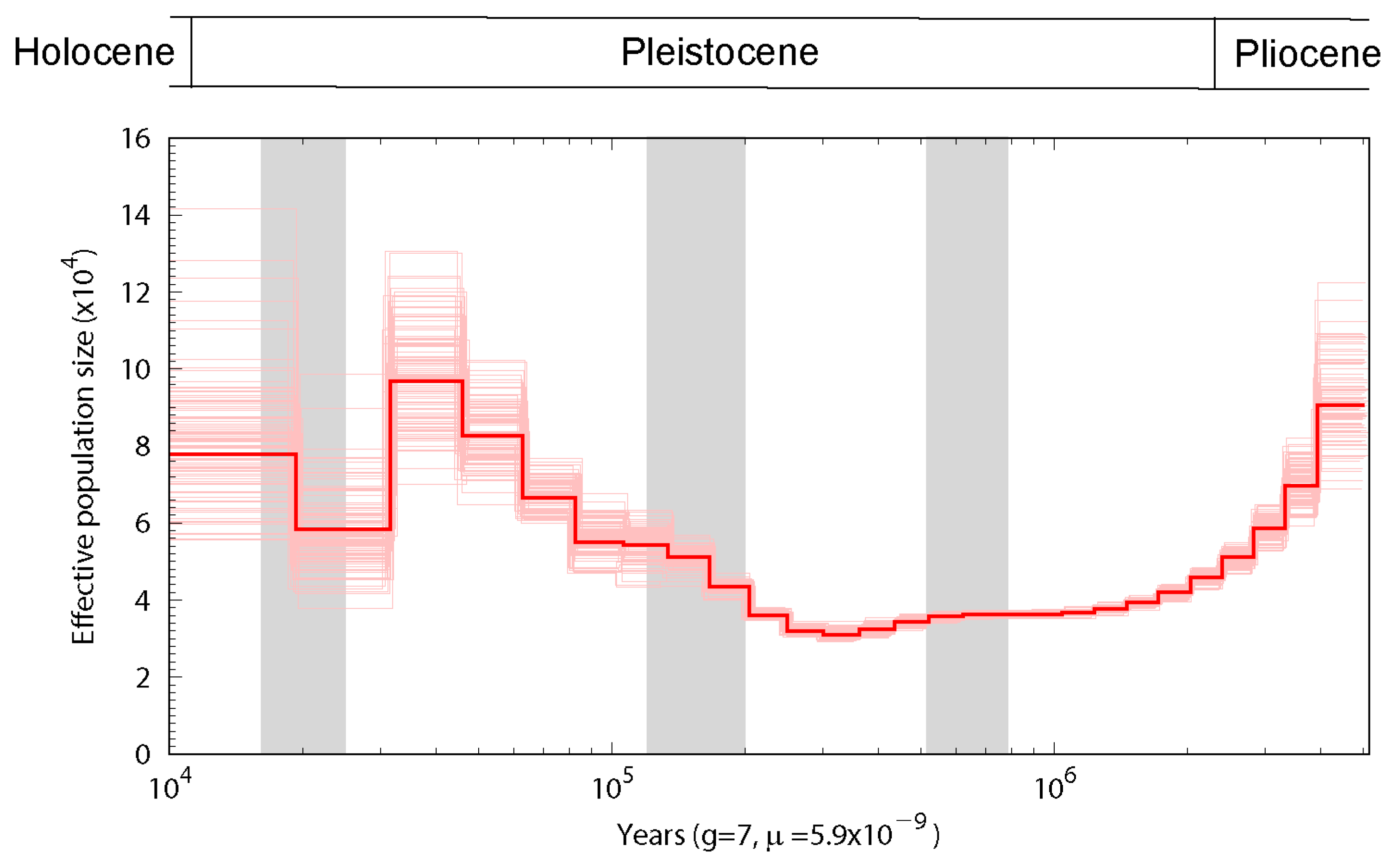
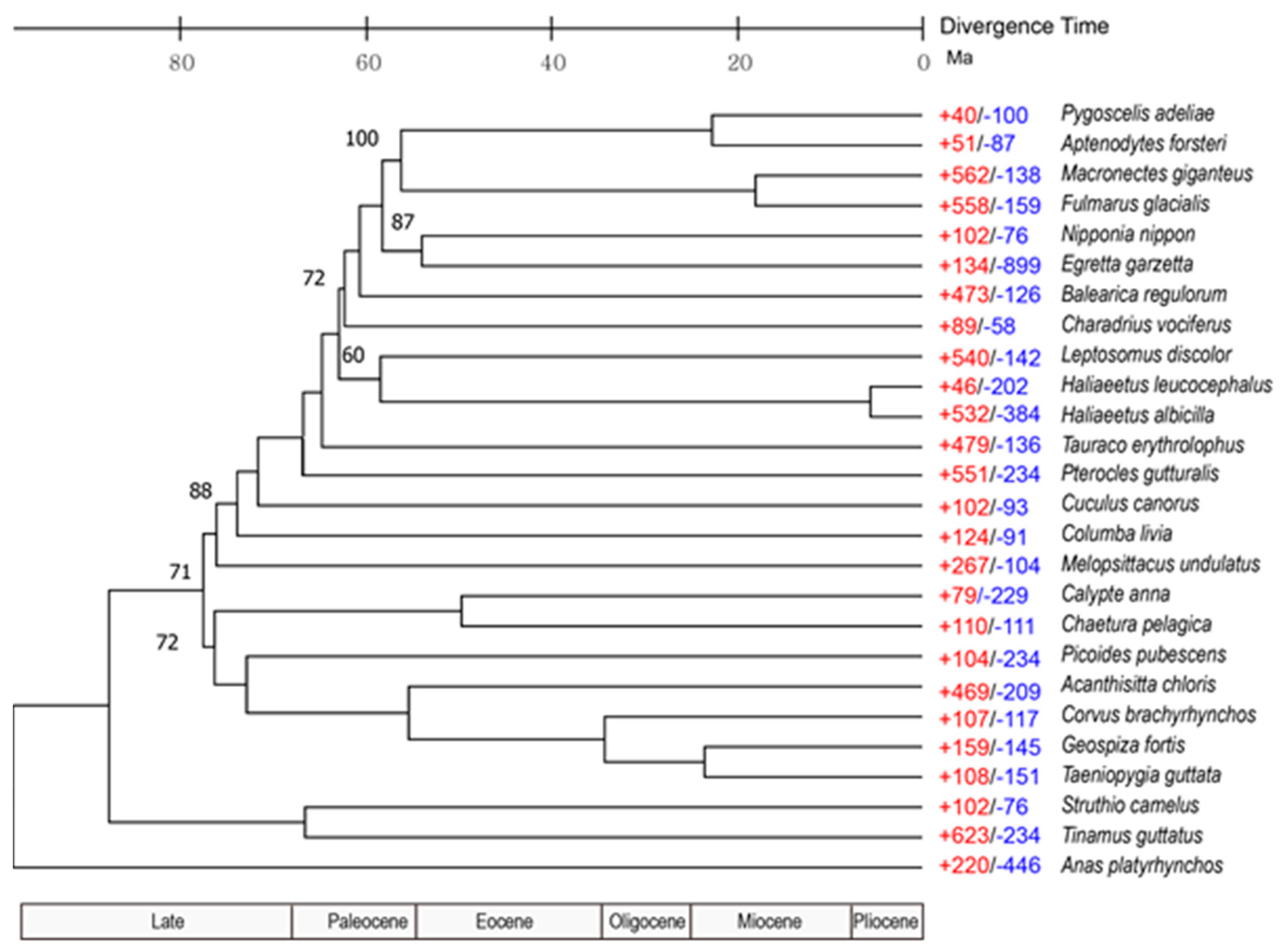
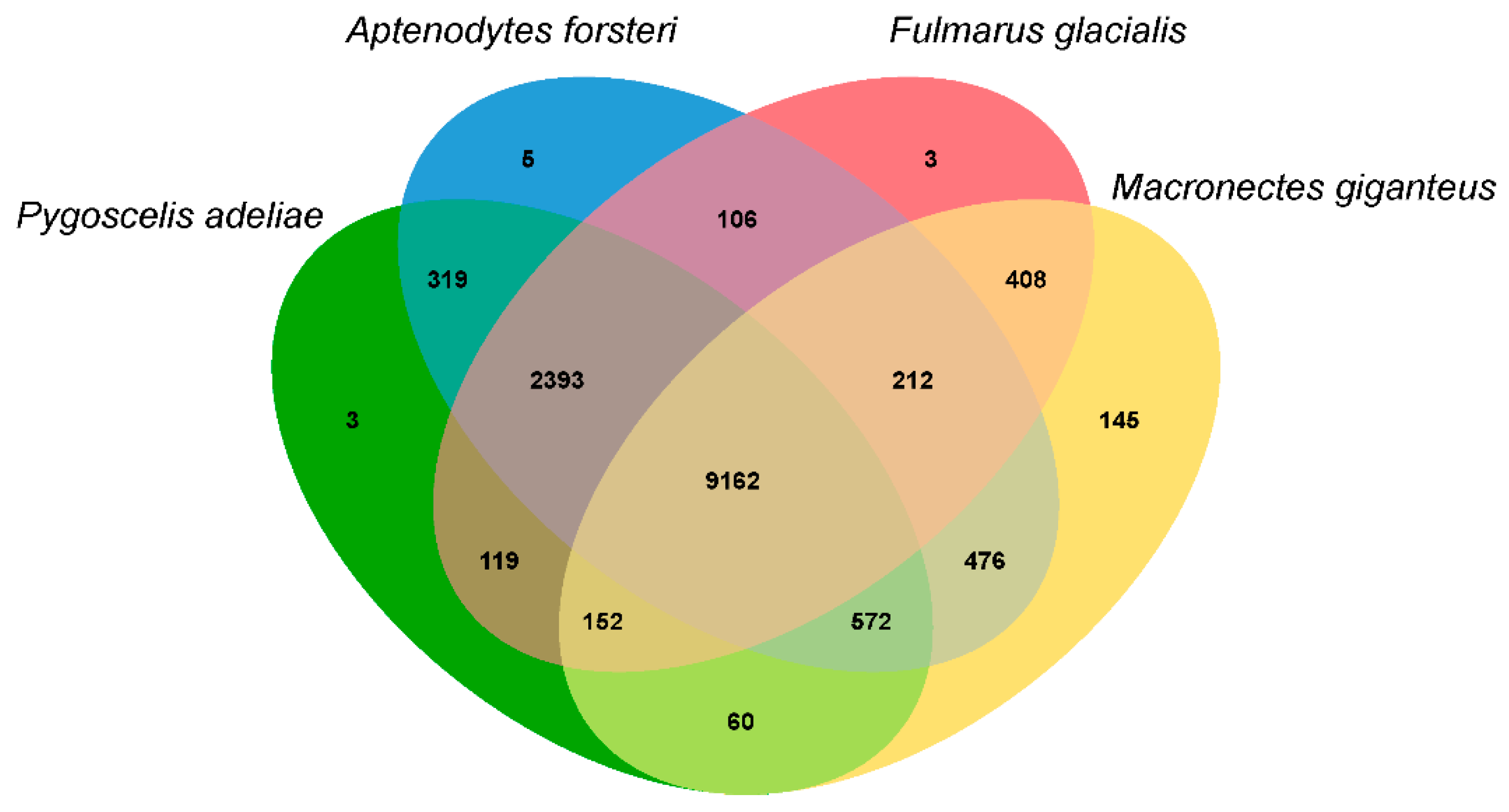
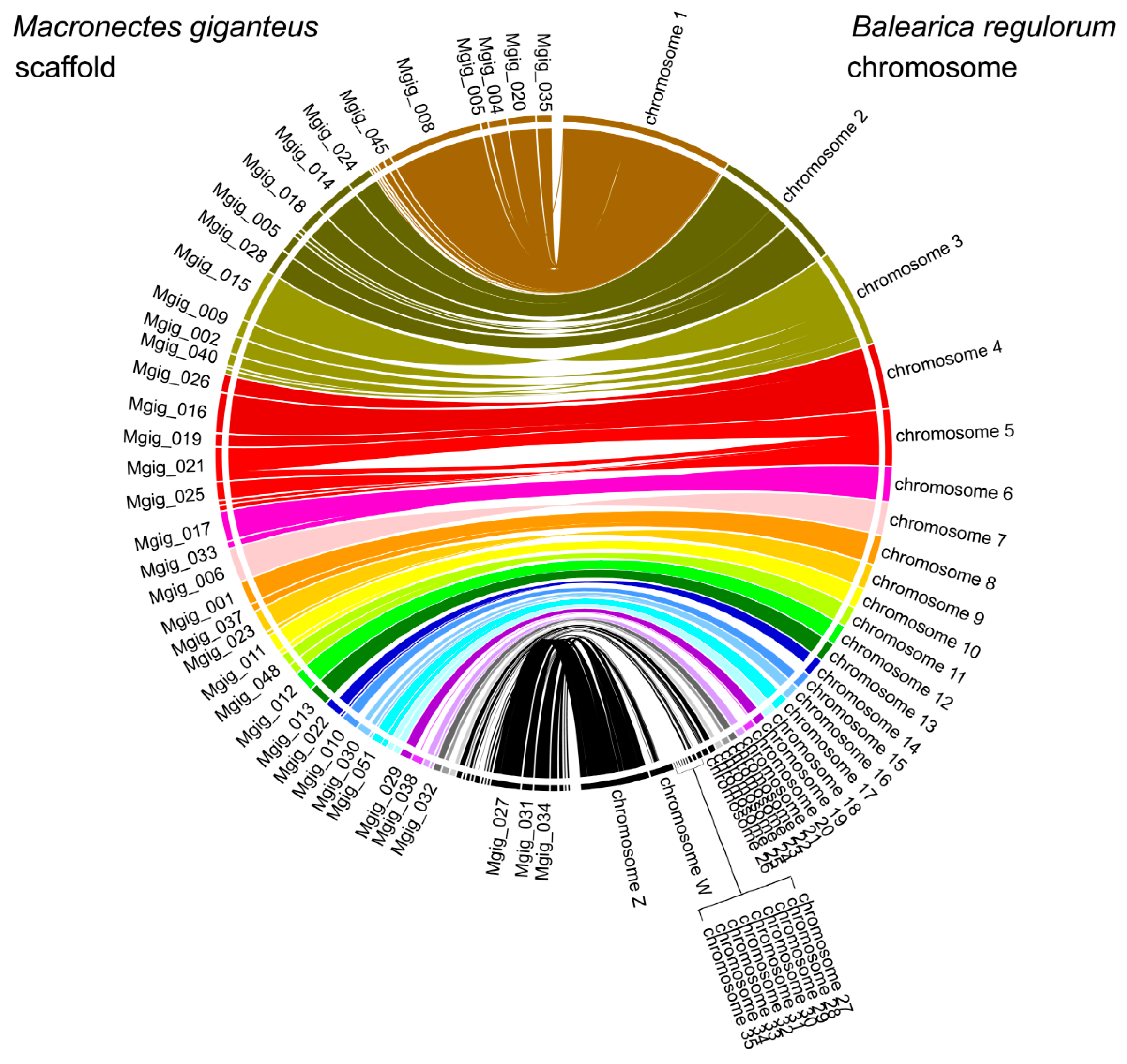
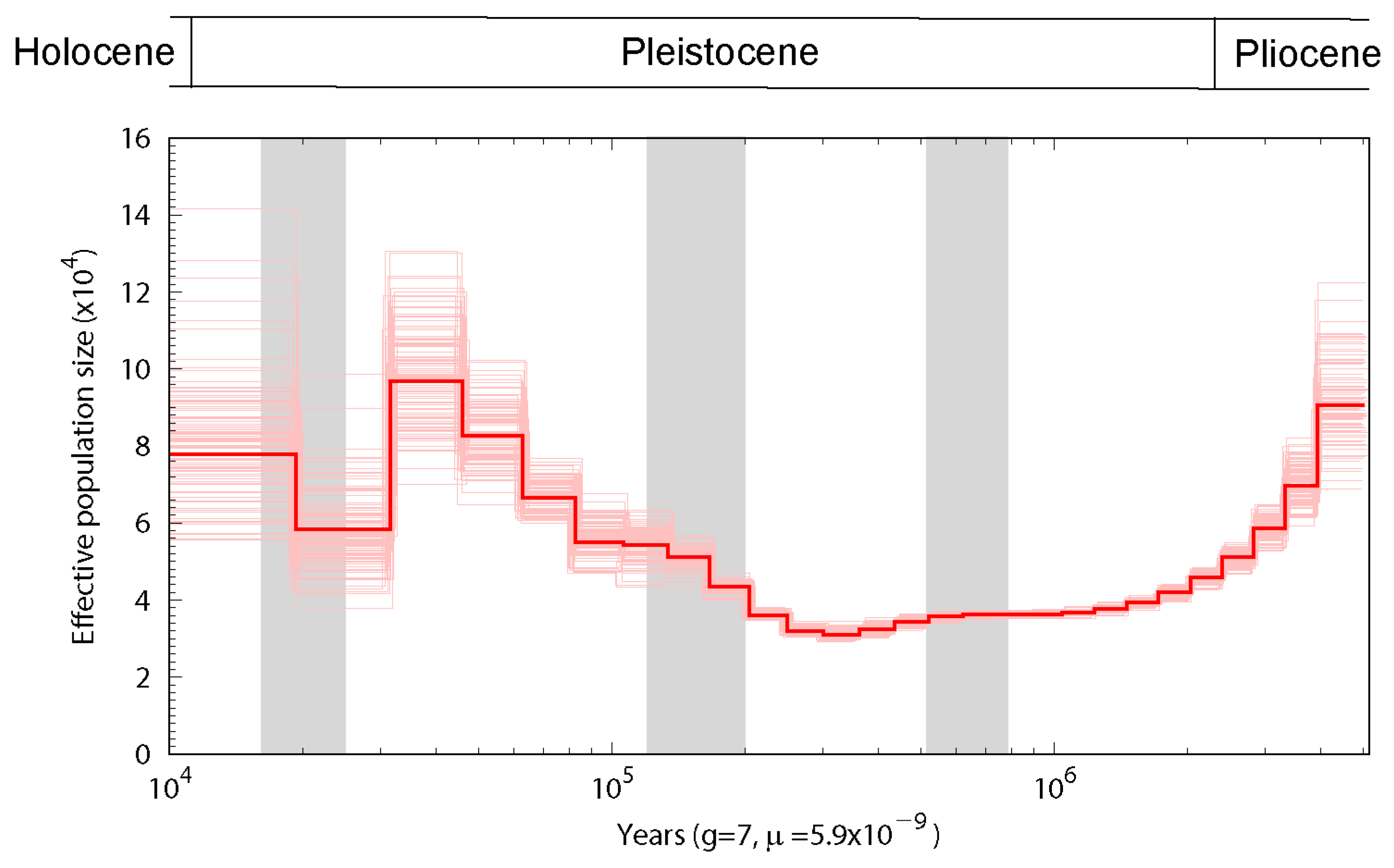
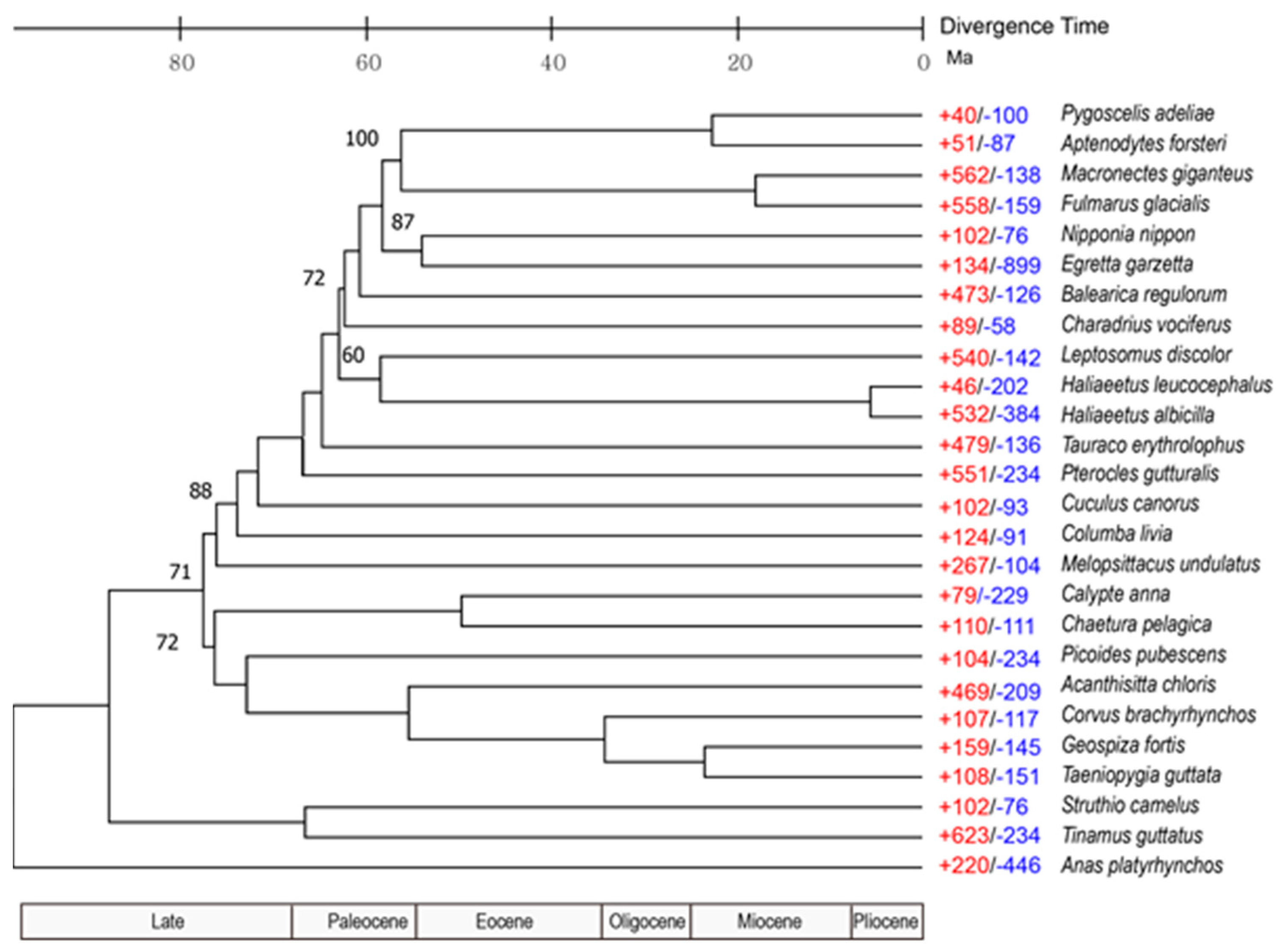
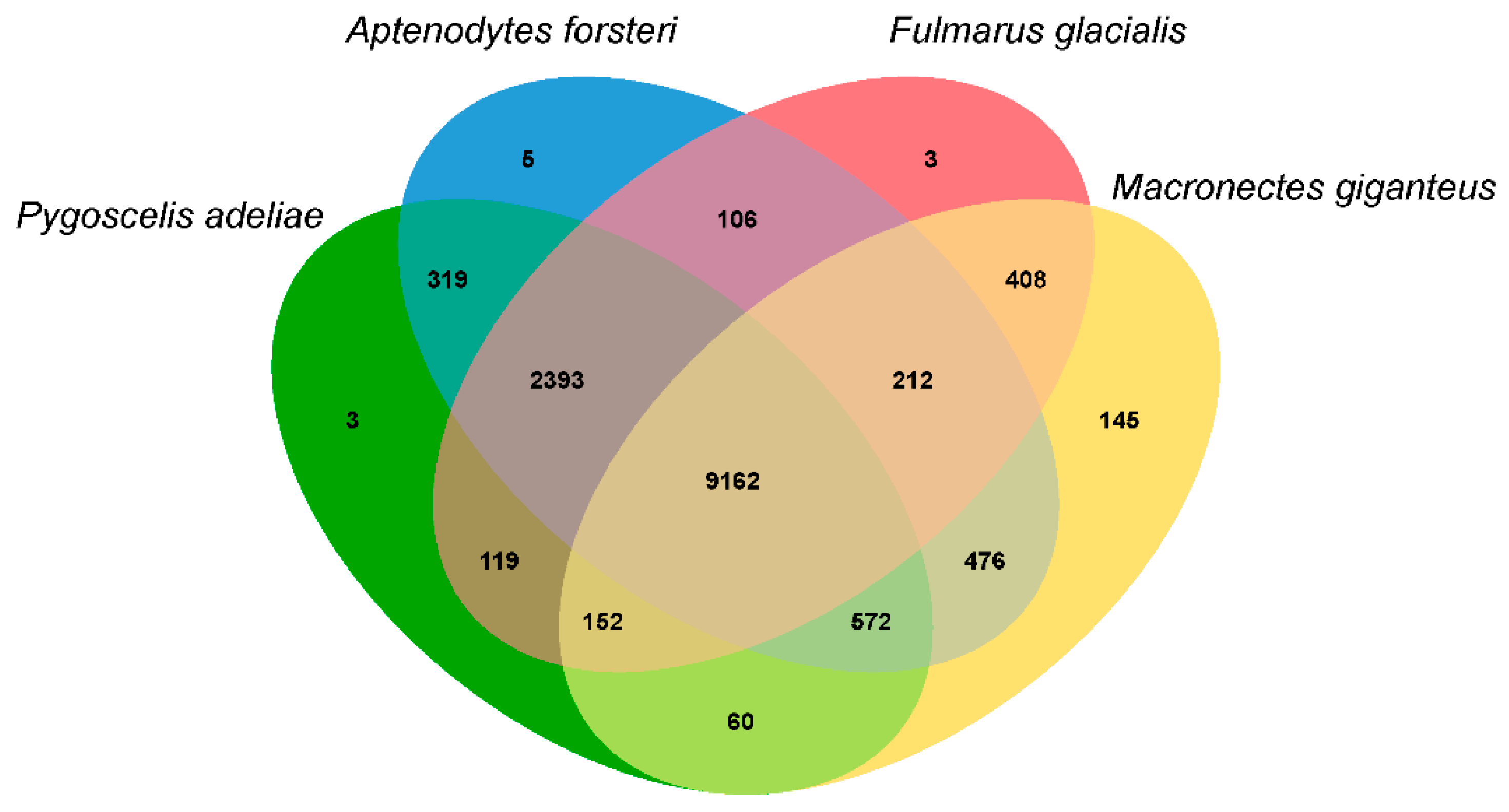
3.3. Population History and Gene Phylogenomics
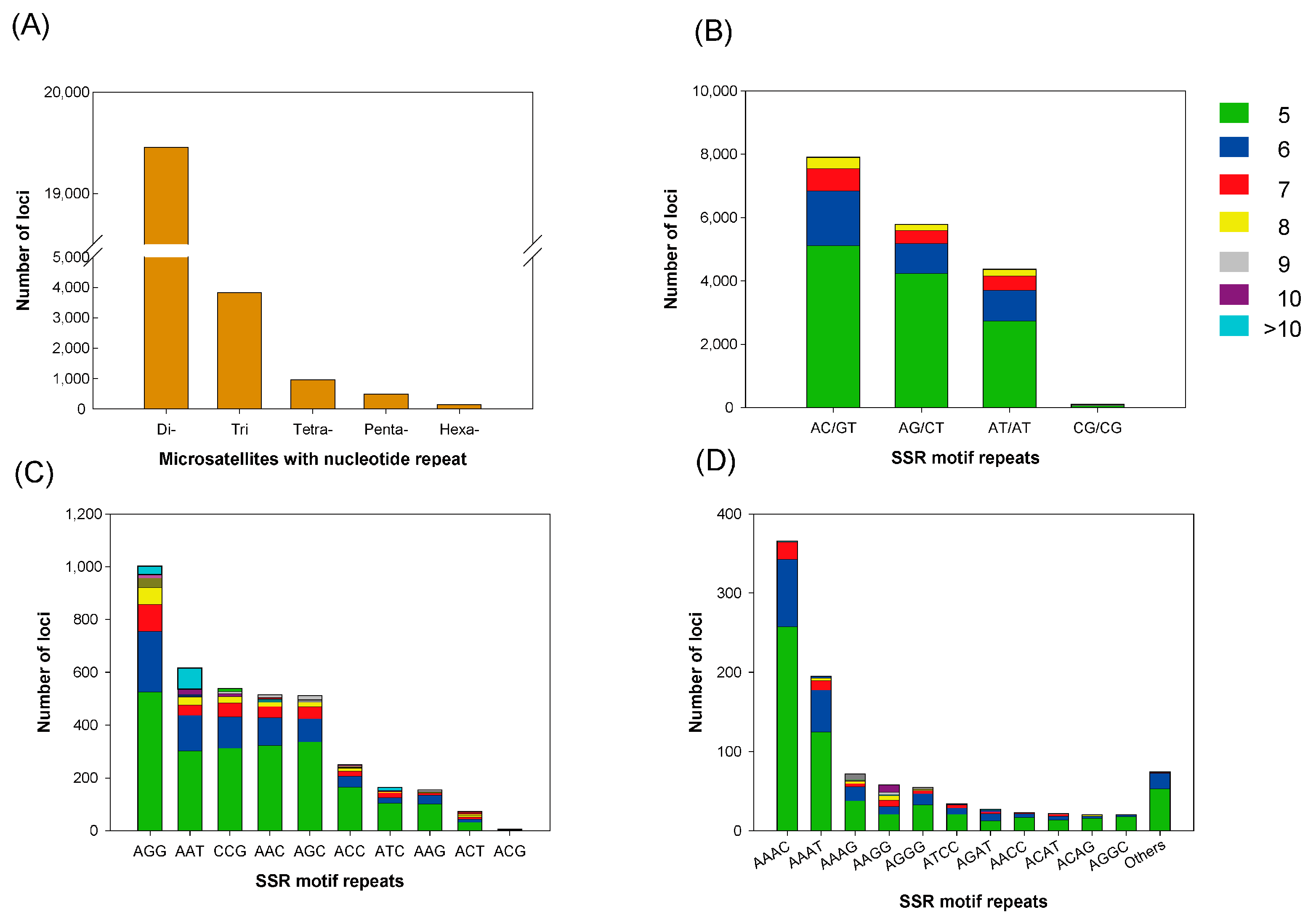
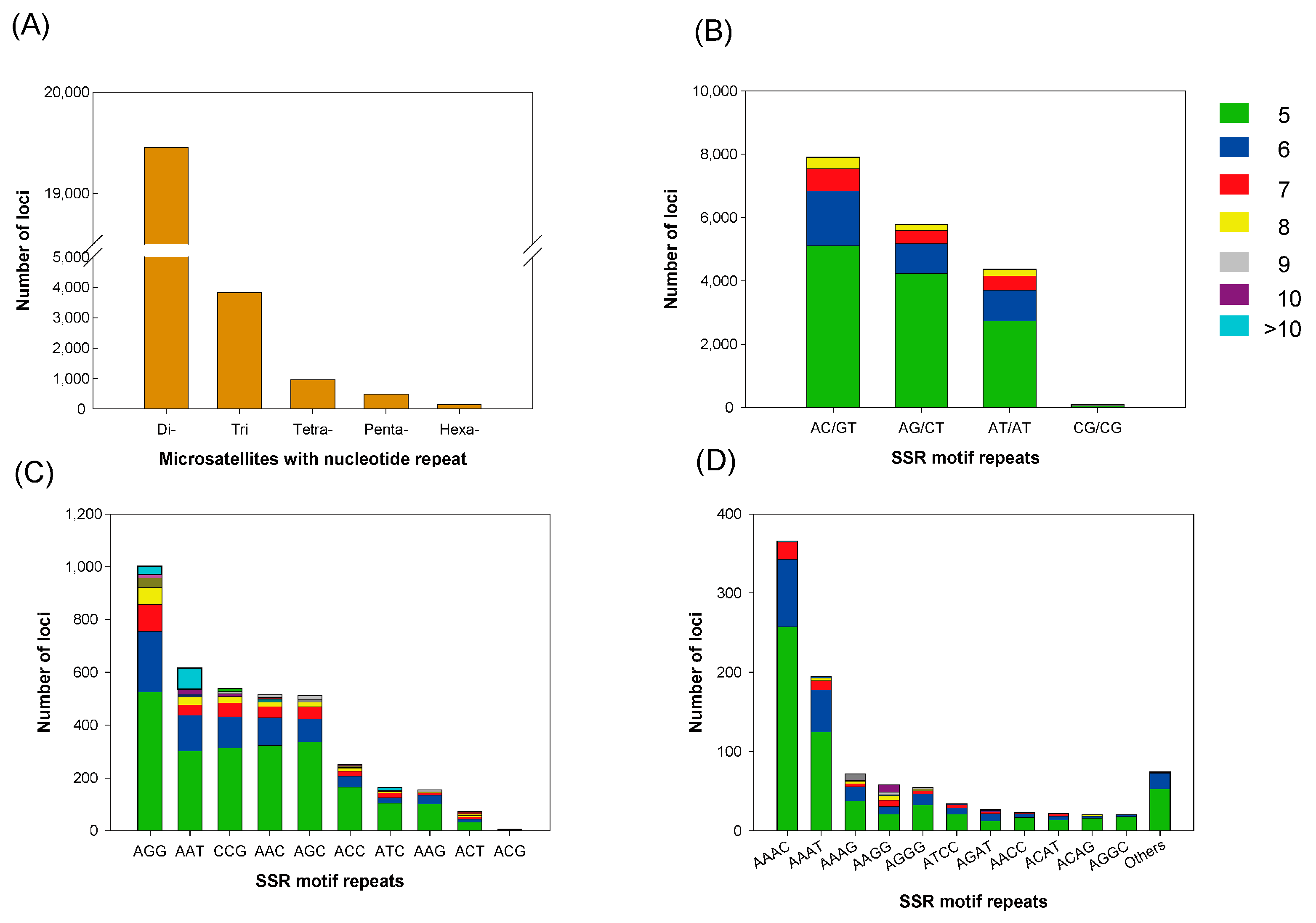
3.4. Microsatellite Marker Identification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bourne, W.R.P.; Warham, J.J.A. Geographical variation in giant petrels of genus macronectes. Ardea 1966, 54, 45. [Google Scholar]
- Brown, R.M.; Mareile Techow, N.M.S.; Wood, A.G.; Phillips, R.A. Hybridization and Back-Crossing in Giant Petrels (Macronectes giganteus and M. halli) at Bird Island, South Georgia, and a Summary of Hybridization in Seabirds. PLoS ONE 2015, 10, e0121688. [Google Scholar] [CrossRef] [Green Version]
- International Union for Conservation of Nature. Macronectes Giganteus; The IUCN Red List of Threatened Species 2018; International Union for Conservation of Nature: Gland, Switzerland, 2018. [Google Scholar]
- Boarman, W.I. Complete Guide to Antarctic Wildlife: Birds and Mammals of the Antarctic Continent and the Southern Ocean. Condor 2004, 106, 447–448. [Google Scholar]
- Johnstone, G.W. Field characters and behaviour at sea of giant petrels in relation to their oceanic distribution. Emu 1974, 74, 209–218. [Google Scholar] [CrossRef]
- Hunter, S. Species and sexual isolating mechanisms in sibling species of giant petrels Macronectes. Polar Biol. 1987, 7, 295–301. [Google Scholar] [CrossRef]
- Johnstone, G.W. Agonistic Behaviour of the Giant-Petrels Macronectes Giganteus and M. Halli Feeding at Seal Carcasses. Emu 1979, 79, 129–132. [Google Scholar] [CrossRef]
- Davies, S. Grzimek’s Animal Life Encyclopedia. 8 Birds I Tinamous and Ratites to Hoatzins; Gale Group: Farmington Hills, MI, USA, 2003. [Google Scholar]
- Vianna, J.A.; Fernandes, F.A.N.; Frugone, M.J.; Figueiró, H.V.; Pertierra, L.R.; Noll, D.; Bi, K.; Wang-Claypool, C.Y.; Lowther, A.; Parker, P.; et al. Genome-wide analyses reveal drivers of penguin diversification. Proc. Natl. Acad. Sci. USA 2020, 117, 22303–22310. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Vurture, G.W.; Sedlazeck, F.; Nattestad, M.; Underwood, C.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: http://www.r-project.org (accessed on 8 July 2021).
- Chin, C.-S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.; Coombe, L.; Warren, R.L.; Chu, J.; Birol, I. ARCS: Scaffolding genome drafts with linked reads. Bioinformatics 2018, 34, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-M.; Ahn, D.-H.; Kim, J.-H.; Jung, J.-W.; Rhee, J.-S.; Park, H. De novo assembly and annotation of the blood transcriptome of the southern giant petrel Macronectes giganteus from the South Shetland Islands, Antarctica. Mar. Genom. 2018, 42, 63–66. [Google Scholar] [CrossRef]
- Eilbeck, K.; Moore, B.; Holt, C.; Yandell, M. Quantitative measures for the management and comparison of annotated genomes. BMC Bioinform. 2009, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinform. 2014, 48, 4.11.11–14.11.39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M.R. Blast2GO: A universal tool for annotation, visu-alization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.I.; Schein, J.E.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An in-formation aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nat. Cell Biol. 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Voisin, J.-F. Breeding biology of the northern giant petrel Macronectes halli and the southern giant petrel M. giganteus at île de la Possession, îles Crozet, 1966-1980. Mar. Ornithol. 1988, 16, 65–97. [Google Scholar]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J., Jr. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinform. 2011, 35, 6.12. 11–16.12. 19. [Google Scholar]
- Löytynoja, A.; Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. USA 2005, 102, 10557–10562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestro, D.; Michalak, I. raxmlGUI: A graphical front-end for RAxML. Org. Divers. Evol. 2012, 12, 335–337. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Bioinformatics 1994, 10, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Meglécz, E.; Pech, N.; Gilles, A.; Dubut, V.; Hingamp, P.; Trilles, A.; Grenier, R.; Martin, J.-F. QDD version 3.1: A user-friendly computer program for microsatellite selection and primer design revisited: Experimental validation of variables determining genotyping success rate. Mol. Ecol. Resour. 2014, 14, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2010, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Nadachowska-Brzyska, K.; Li, C.; Smeds, L.; Zhang, G.; Ellegren, H. Temporal Dynamics of Avian Populations during Pleistocene Revealed by Whole-Genome Sequences. Curr. Biol. 2015, 25, 1375–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, D.L.; Woehler, E.J.; Croxall, J.P.; Cooper, J.; Poncet, S.; Hunter, S.; Fraser, W.R. Breeding distribution and population status of the Northern Giant Petrel Macronectes halli and Southern Giant Petrel M. giganteus. Mar. Ornithol. 2008, 36, 115–124. [Google Scholar]
- Brooke, M. Albatrosses and Petrels Across the World; Oxford University Press: Oxford, UK, 2004. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contigs (Falcon-Unzip) | Scaffolds (ARCS) | |
|---|---|---|
| Number of scaffolds | 1199 | 861 |
| Total size of scaffolds | 1,247,954,784 | 1,247,958,164 |
| Longest scaffold | 20,833,995 | 120,319,780 |
| Shortest scaffold | 15,076 | 15,076 |
| Number of scaffolds >10 M nt | 21 | 36 |
| N50 scaffold length | 6,274,151 | 27,376,298 |
| L50 scaffold number | 61 | 14 |
| G+C content (%) | 42.76 | 42.76 |
| aves_odb9 | No. | % |
|---|---|---|
| Complete BUSCOs (C) | 4627 | 94.2 |
| Complete and single-copy BUSCOs (S) | 4570 | 93 |
| Complete and duplicated BUSCOs (D) | 57 | 1.2 |
| Fragmented BUSCOs (F) | 183 | 3.7 |
| Missing BUSCOs (M) | 105 | 2.1 |
| Total BUSCO groups searched | 4915 |
| Value | |
|---|---|
| Gene length sum (bp) | 269,696,814 |
| Gene count | 14,993 |
| Exon length sum (bp) | 27,620,459 |
| Exon count | 152,053 |
| CDS length sum (bp) | 25,123,992 |
| CDS count | 151,205 |
| NCBI-NR | 14,779 |
| Uniprot/Swiss-prot annotations | 10,034 |
| InterProScan annotations | 14,764 |
| Pfam annotations | 10,033 |
| EggNOG annotations | 12,665 |
| Gene Ontology annotations | 12,562 |
| KEGG annotations | 9154 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.-H.; Lee, S.-J.; Jo, E.; Kim, J.; Kim, J.-U.; Kim, J.-H.; Park, H.; Chi, Y.-M. Genome of the Southern Giant Petrel Assembled Using Third-Generation DNA Sequencing and Linked Reads Reveals Evolutionary Traits of Southern Avian. Animals 2021, 11, 2046. https://doi.org/10.3390/ani11072046
Kim S-H, Lee S-J, Jo E, Kim J, Kim J-U, Kim J-H, Park H, Chi Y-M. Genome of the Southern Giant Petrel Assembled Using Third-Generation DNA Sequencing and Linked Reads Reveals Evolutionary Traits of Southern Avian. Animals. 2021; 11(7):2046. https://doi.org/10.3390/ani11072046
Chicago/Turabian StyleKim, Sun-Hee, Seung-Jae Lee, Euna Jo, Jangyeon Kim, Jong-U Kim, Jeong-Hoon Kim, Hyun Park, and Young-Min Chi. 2021. "Genome of the Southern Giant Petrel Assembled Using Third-Generation DNA Sequencing and Linked Reads Reveals Evolutionary Traits of Southern Avian" Animals 11, no. 7: 2046. https://doi.org/10.3390/ani11072046
APA StyleKim, S.-H., Lee, S.-J., Jo, E., Kim, J., Kim, J.-U., Kim, J.-H., Park, H., & Chi, Y.-M. (2021). Genome of the Southern Giant Petrel Assembled Using Third-Generation DNA Sequencing and Linked Reads Reveals Evolutionary Traits of Southern Avian. Animals, 11(7), 2046. https://doi.org/10.3390/ani11072046