
Investigating the Effects of a Phytobiotics-Based Product on the Fecal Bacterial Microbiome of Weaned Pigs




Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Management and Sample Collection
2.2. Isolation of Microbial DNA and 16S rRNA Gene Amplification by PCR
2.3. Bacterial Composition Analyses
2.4. Statistical Analyses
2.5. Next Generation Sequencing Data Accessibility
3. Results
3.1. Pig Growth and Fecal Scores
3.2. Taxonomic Composition Analysis of Fecal Bacterial Communities
3.3. OTU Composition Analysis of Fecal Bacterial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Amezcua, R.; Friendship, R.M.; Dewey, C.E.; Gyles, C.; Fairbrother, J.M. Presentation of postweaning Escherichia coli diarrhea in southern Ontario, prevalence of hemolytic E. coli serogroups involved, and their antimicrobial resistance patterns. Can. J. Vet. Res. 2002, 66, 73. [Google Scholar]
- Fairbrother, J.M.; Nadeau, É.; Gyles, C.L. Escherichia coli in postweaning diarrhea in pigs: An update on bacterial types, pathogenesis, and prevention strategies. Anim. Health Res. Rev. 2005, 6, 17. [Google Scholar] [CrossRef]
- Luppi, A.; Gibellini, M.; Gin, T.; Vangroenweghe, F.; Vandenbroucke, V.; Bauerfeind, R.; Bonilauri, P.; Labarque, G.; Hidalgo, Á. Prevalence of virulence factors in enterotoxigenic Escherichia coli isolated from pigs with post-weaning diarrhoea in Europe. Porc. Health Manag. 2016, 2, 1–6. [Google Scholar] [CrossRef]
- Sun, Y.; Kim, S.W. Intestinal challenge with enterotoxigenic Escherichia coli in pigs, and nutritional intervention to prevent postweaning diarrhea. Anim. Nutr. 2017, 3, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Windisch, W.; Schedle, K.; Plitzner, C.; Kroismayr, A. Use of phytogenic products as feed additives for swine and poultry. J. Anim. Sci. 2008, 86, E140–E148. [Google Scholar] [CrossRef] [PubMed]
- Kroismayr, A.; Sehm, J.; Pfaffl, M.; Plitzner, C.; Foissy, H.; Ettle, T.; Mayer, H.; Schreiner, M.; Windisch, W. Effects of essential oils or avilamycin on gut physiology and blood parameters of weaned piglets. Czech. J. Anim. Sci. 2008, 53, 377–387. [Google Scholar] [CrossRef]
- Manzanilla, E.G.; Perez, J.F.; Martin, M.; Kamel, C.; Baucells, F.; Gasa, J. Effect of plant extracts and formic acid on the intestinal equilibrium of early-weaned pigs1. J. Anim. Sci. 2004, 82, 3210–3218. [Google Scholar] [CrossRef]
- Castillo, M.; Martín-Orúe, S.M.; Roca, M.; Manzanilla, E.G.; Badiola, I.; Perez, J.F.; Gasa, J. The response of gastrointestinal microbiota to avilamycin, butyrate, and plant extracts in early-weaned pigs1,2. J. Anim. Sci. 2006, 84, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Morrison, M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 2004, 36, 808–812. [Google Scholar] [CrossRef]
- Edwards, U.; Rogall, T.; Blöcker, H.; Emde, M.; Böttger, E.C. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989, 17, 7843–7853. [Google Scholar] [CrossRef]
- Lane, D.J.; Pace, B.; Olsen, G.J.; Stahl, D.A.; Sogin, M.L.; Pace, N.R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA 1985, 82, 6955–6959. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Weber, C.F. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 423, 75. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Morrison, M.; Yu, Z. Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J. Microbiol. Methods 2011, 84, 81–87. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Parte, A.C. LPSN—list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014, 42, D613–D616. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community Ecol. Package Version 2013, 2, 1–295. [Google Scholar]
- Campbell, J.M.; Crenshaw, J.D.; Polo, J. The biological stress of early weaned piglets. J. Anim. Sci. Biotechnol. 2013, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Messer, J.S.; Chang, E.B. Chapter 36—Microbial physiology of the digestive tract and its role in inflammatory bowel diseases. In Physiology of the Gastrointestinal Tract, 6th ed.; Said, H.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 795–810. [Google Scholar]
- Gresse, R.; Chaucheyras-Durand, F.; Fleury, M.A.; Van de Wiele, T.; Forano, E.; Blanquet-Diot, S. Gut microbiota dysbiosis in postweaning piglets: Understanding the keys to health. Trends Microbiol. 2017, 25, 851–873. [Google Scholar] [CrossRef] [PubMed]
- Bontempo, V.; Jiang, X.; Cheli, F.; Lo Verso, L.; Mantovani, G.; Vitari, F.; Domeneghini, C.; Agazzi, A. Administration of a novel plant extract product via drinking water to post-weaning piglets: Effects on performance and gut health. Animal 2014, 8, 721–730. [Google Scholar] [CrossRef]
- Wu, C.; Wu, G. A novel plant extract mix, GrazixTM, is capable of binding endotoxin. In Proceedings of the 4th International Feed Safety Conference, Beijing, China, 11–13 September 2012; pp. 11–13. [Google Scholar]
- Wenk, C. Herbs and botanicals as feed additives in monogastric animals. Asian Australas J. Anim. Sci. 2003, 16, 282–289. [Google Scholar] [CrossRef]
- Li, P.; Piao, X.; Ru, Y.; Han, X.; Xue, L.; Zhang, H. Effects of adding essential oil to the diet of weaned pigs on performance, nutrient utilization, immune response and intestinal health. Asian Australas. J. Anim. Sci. 2012, 25, 1617. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Gong, J.; Tsao, R.; Zhou, T.; Yu, H.; Poppe, C.; Johnson, R.; Du, Z. Antimicrobial activity of essential oils and structurally related synthetic food additives towards selected pathogenic and beneficial gut bacteria. J. Appl. Microbiol. 2006, 100, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.; Missotten, J.; Fremaut, D.; De Smet, S.; Dierick, N. In vitro characterisation of the antimicrobial activity of selected essential oil components and binary combinations against the pig gut flora. Anim. Feed Sci. Technol. 2009, 151, 111–127. [Google Scholar] [CrossRef]
- Burt, S. Essential oils: Their antibacterial properties and potential applications in foods—A review. Int. J. Food Microbiol. 2004, 94, 223–253. [Google Scholar] [CrossRef]
- Hu, J.; Nie, Y.; Chen, J.; Zhang, Y.; Wang, Z.; Fan, Q.; Yan, X. Gradual changes of gut microbiota in weaned miniature piglets. Front. Microbiol. 2016, 7, 1727. [Google Scholar] [CrossRef]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 2011, 153, 124–133. [Google Scholar] [CrossRef]
- Poudel, P.; Levesque, C.L.; Samuel, R.; St-Pierre, B. Dietary inclusion of Peptiva, a peptide-based feed additive, can accelerate the maturation of the fecal bacterial microbiome in weaned pigs. BMC Vet. Res. 2020, 16, 1–13. [Google Scholar] [CrossRef]
- Yang, F.; Hou, C.; Zeng, X.; Qiao, S. The use of lactic acid bacteria as a probiotic in swine diets. Pathogens 2015, 4, 34–45. [Google Scholar] [CrossRef]
- Vieco-Saiz, N.; Belguesmia, Y.; Raspoet, R.; Auclair, E.; Gancel, F.; Kempf, I.; Drider, D. Benefits and inputs from lactic acid bacteria and their bacteriocins as alternatives to antibiotic growth promoters during food-animal production. Front. Microbiol. 2019, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, R.; Yocheva, L.; Tserovska, L.; Zhelezova, G.; Stefanova, N.; Atanasova, A.; Danguleva, A.; Ivanova, G.; Karapetkov, N.; Rumyan, N. Antimicrobial activity and antibiotic susceptibility of Lactobacillus and Bifidobacterium spp. intended for use as starter and probiotic cultures. Biotechnol. Biotechnol. Equip. 2015, 29, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Inglin, R.C.; Stevens, M.J.; Meile, L.; Lacroix, C.; Meile, L. High-throughput screening assays for antibacterial and antifungal activities of Lactobacillus species. J. Microbiol. Methods 2015, 114, 26–29. [Google Scholar] [CrossRef]
- Fouhse, J.M.; Zijlstra, R.T.; Willing, B.P. The role of gut microbiota in the health and disease of pigs. Anim. Front. 2016, 6, 30–36. [Google Scholar] [CrossRef]
- Huang, C.; Qiao, S.; Li, D.; Piao, X.; Ren, J. Effects of Lactobacilli on the performance, diarrhea incidence, VFA concentration and gastrointestinal microbial flora of weaning pigs. Asian Australas J. Anim Sci. 2004, 17, 401–409. [Google Scholar] [CrossRef]
- Valeriano, V.D.; Balolong, M.P.; Kang, D.K. Probiotic roles of Lactobacillus sp. in swine: Insights from gut microbiota. J. Appl. Microbiol. 2007, 122, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, L. Lactobacillus amylovorus, a new starch-hydrolyzing species from cattle waste-corn fermentations. Int. J. Syst. Evol. Microbiol. 1981, 31, 56–63. [Google Scholar] [CrossRef]
- Hammes, W.P.; Hertel, C. The genera Lactobacillus and Carnobacterium. Prokaryotes 2006, 4, 320–403. [Google Scholar]
- Chang, Y.-H.; Kim, J.-K.; Kim, H.-J.; Kim, W.-Y.; Kim, Y.-B.; Park, Y.-H. Selection of a potential probiotic Lactobacillus strain and subsequent in vivo studies. Antonie Van Leeuwenhoek 2001, 80, 193–199. [Google Scholar] [CrossRef]
- Su, Y.; Li, B.; Zhu, W.-Y. Fecal microbiota of piglets prefer utilizing dl-lactate mixture as compared to d-lactate and l-lactate in vitro. Anaerobe 2013, 19, 27–33. [Google Scholar] [CrossRef]
- Ortman, J.; Sinn, S.; Gibbons, W.; Brown, M.; DeRouchey, J.; St-Pierre, B.; Saqui-Salces, M.; Levesque, C. Comparative analysis of the ileal bacterial composition of post-weaned pigs fed different high-quality protein sources. Animal 2020, 14, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Kant, R.; Paulin, L.; Alatalo, E.; de Vos, W.M.; Palva, A. Genome sequence of Lactobacillus amylovorus GRL1118, isolated from pig ileum. J. Bacteriol. 2011, 193, 3147–3148. [Google Scholar] [CrossRef]
- Pajarillo, E.A.B.; Chae, J.-P.; Balolong, M.P.; Kim, H.B.; Kang, D.-K. Assessment of fecal bacterial diversity among healthy piglets during the weaning transition. J. Gen. Appl. Microbiol. 2014, 60, 140–146. [Google Scholar] [CrossRef]
- Niu, Q.; Li, P.; Hao, S.; Zhang, Y.; Kim, S.W.; Li, H.; Ma, X.; Gao, S.; He, L.; Wu, W. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 2015, 5, 9938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, Y.; Liu, S.; Huang, J.; Zhai, Z.; He, C.; Ding, J.; Wang, J.; Wang, H.; Fan, W. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 2015, 10, e0117441. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Ewaschuk, J.B.; Naylor, J.M.; Zello, G.A. D-lactate in human and ruminant metabolism. J. Nutr. 2005, 135, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. D-lactic acid as a metabolite: Toxicology, diagnosis, and detection. BioMed Res. Int. 2020, 2020, 3419034. [Google Scholar] [CrossRef]
- Nabuurs, M.; Van Essen, G.; Nabuurs, P.; Niewold, T.; Van Der Meulen, J. Thirty minutes transport causes small intestinal acidosis in pigs. Res. Vet. Sci. 2001, 70, 123–127. [Google Scholar] [CrossRef]
- Van Beers-Schreurs, H.; Vellenga, L.; Wensing, T.; Breukink, H. The pathogenesis of the post-weaning syndrome in weaned piglets; a review. Vet. Q. 1992, 14, 29–34. [Google Scholar] [CrossRef]
- Counotte, G.H.; Prins, R.A.; Janssen, R.H.; Debie, M.J. Role of Megasphaera elsdenii in the fermentation of dl-[2-C]lactate in the rumen of dairy cattle. Appl. Environ. Microbiol. 1981, 42, 649–655. [Google Scholar] [CrossRef]
- Patterson, M.J. Streptococcus. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; Chapter 13. [Google Scholar]
- Schlegel, L.; Grimont, F.; Ageron, E.; Grimont, P.A.; Bouvet, A. Reappraisal of the taxonomy of the Streptococcus bovis/Streptococcus equinus complex and related species: Description of Streptococcus gallolyticus subsp. gallolyticus subsp. nov., S. gallolyticus subsp. macedonicus subsp. nov. and S. gallolyticus subsp. pasteurianus subsp. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 631–645. [Google Scholar]
- Farrow, J.; Kruze, J.; Phillips, B.; Bramley, A.; Collins, M. Taxonomic studies on Streptococcus bovis and Streptococcus equinus: Description of Streptococcus alactolyticus sp. nov. and Streptococcus saccharolyticus sp. nov. Syst. Appl. Microbiol. 1984, 5, 467–482. [Google Scholar] [CrossRef]
- Rinkinen, M.L.; Koort, J.M.K.; Ouwehand, A.C.; Westermarck, E.; Björkroth, K.J. Streptococcus alactolyticus is the dominating culturable lactic acid bacterium species in canine jejunum and feces of four fistulated dogs. FEMS Microbiol. Lett. 2004, 230, 35–39. [Google Scholar] [CrossRef]
- Robinson, I.; Stromley, J.; Varel, V.; Cato, E. Streptococcus intestinalis, a new species from the colons and feces of pigs. Int. J. Syst. Evol. Microbiol. 1988, 38, 245–248. [Google Scholar] [CrossRef]
- Chun, C.; Zheng, L.; Colgan, S.P. Tissue metabolism and host-microbial interactions in the intestinal mucosa. Free Radic. Biol. Med. 2017, 105, 86–92. [Google Scholar] [CrossRef]
- Canani, R.B.; Di Costanzo, M.; Leone, L. The epigenetic effects of butyrate: Potential therapeutic implications for clinical practice. Clin. Epigenetics 2012, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Zeamer, K.M.; Samuel, R.S.; Pierre, B.S.; Thaler, R.C.; Woyengo, T.A.; Hymowitz, T.; Levesque, C.L. Effects of a low allergenic soybean variety on gut permeability, microbiota composition, ileal digestibility of amino acids, and growth performance in pigs. Livest. Sci. 2020, 243, 104369. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
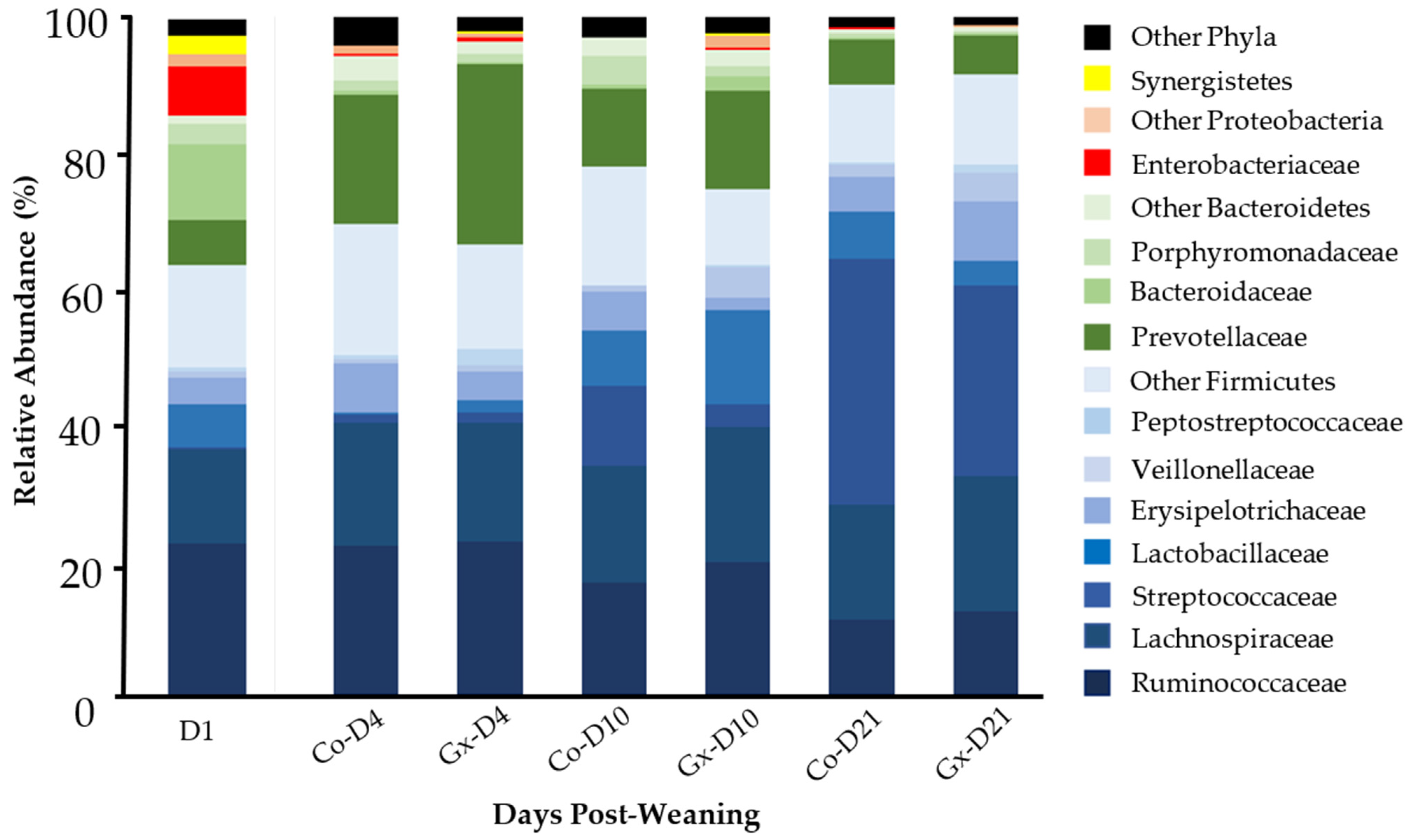
| Taxon | D1 | Co-D4 | Gx D4 | Co-D10 | Gx D10 | Co-D21 | Gx D21 |
|---|---|---|---|---|---|---|---|
| Firmicutes # | 63.8 a | 69.6 ab | 66.6 ab | 78.1 b | 74.8 b | 90.1 c | 91.5 c |
| Ruminococcaceae # | 22.7 a | 22.2 a | 22.7 a | 16.8 ab | 19.6 ab | 11.2 b | 12.3 b |
| Lachnospiraceae | 13.8 | 18.0 | 17.4 | 17.1 | 20.0 | 16.9 | 20.0 |
| Streptococcaceae # | 0.5 a | 1.2 ab | 1.8 b | 11.8 c | 3.4 b | 36.4 d | 28.3 e |
| Erysipelotrichaceae # | 4.0 ab | 7.3 a | 4.4 a | 5.8 a | 2.0 b | 5.1 a | 8.6 a |
| Lactobacillaceae # | 6.2 a | 0.3 c | 1.6 b | 8.2 ab | 13.8 ab | 7.0 a | 3.6 a |
| Veillonellaceae # | 0.9 a | 0.5 a | 0.9 a | 0.7 a | 4.4 b | 1.6 a | 4.4 b |
| Clostridiaceae1# | 0.6 a | 7.9 b | 7.6 b | 6.5 bc | 1.2 ac | 1.5 c | 3.0 abc |
| Peptostreptococcaceae # | 0.6 a | 0.7 c | 2.2 b | 0.2 d | 0.2 ad | 0.3 ad | 1.1 abcd |
| Eubacteriaceae # | 2.9 a | 0.2 abc | 0.2 bc | 0.1 c | 0.5 abc | 0.6 a | 0.4 ab |
| Clostridiales Inc. Sedis_XIII # | 1.0 abc | 1.0 a | 0.8 abc | 1.0 a | 1.3 ab | 0.4 bc | 0.3 c |
| Other Firmicutes $ | 10.6 | 10.4 | 7.1 | 10.1 | 8.5 | 9.1 | 9.6 |
| Bacteroidetes # | 22.2 a | 24.8 a | 30.0 a | 18.9 a | 20.6 a | 8.3 b | 7.0 b |
| Porphyromonadaceae # | 2.8 ab | 1.6 ab | 1.2 ad | 4.3 b | 1.7 ab | 0.6 cd | 0.4 c |
| Prevotellaceae # | 6.7 a | 18.9 bc | 26.5 b | 11.3 ac | 14.4 bcd | 6.8 ad | 6.0 a |
| Bacteroidaceae # | 11.3 a | 0.6 bd | 0.5 bd | 0.6 bd | 2.0 b | 0.2 cd | 0.2 c |
| Other Bacteroidetes $ | 1.36 | 3.7 | 1.7 | 2.7 | 2.6 | 0.6 | 0.5 |
| Proteobacteri # | 8.9 a | 1.5 bd | 1.2 b | 0.2 cd | 2.1 b | 0.2 c | 0.4 d |
| Enterobacteriaceae # | 7.1 a | 0.3 bc | 0.5 b | <0.1 de | <0.1 c | <0.1 d | <0.1 ce |
| Other Proteobacteria $ | 1.8 | 1.2 | 0.7 | 0.2 | 2.0 | 0.2 | 0.3 |
| Synergistetes # | 2.8 a | 0.1 b | <0.1 b | 0 *c | <0.1 c | <0.1 c | 0 *c |
| Other Bacteria $ | 2.3 | 4.0 | 2.2 | 2.9 | 2.4 | 1.6 | 1.2 |
| Index | D1 | Co-D4 | Gx D4 | Co-D10 | Gx D10 | Co-D21 | Gx D21 | p |
|---|---|---|---|---|---|---|---|---|
| Observed OTUs | 412 a | 722 b | 687 bc | 620 bc | 692 b | 485 ac | 519 abc | <0.001 |
| Chao | 625 a | 1217 b | 1249 b | 1088 bc | 1240 bc | 798 a | 893 abc | <0.001 |
| Ace | 789 a | 1679 b | 1768 b | 1448 bc | 1772 bc | 1089 a | 1240 abc | <0.001 |
| Shannon | 4.26 ab | 5.07 c | 4.83 ac | 4.74 abc | 4.88 ac | 4.03 b | 4.32 abc | <0.001 |
| Simpson | 0.06 a | 0.03 a | 0.05 a | 0.04 a | 0.04 a | 0.10 b | 0.07 ab | 0.0016 |
| OTUs | D1 | Co-D4 | Gx D4 | Co-D10 | Gx D10 | Co-D21 | Gx D21 |
|---|---|---|---|---|---|---|---|
| Ssd-00001 | 1.50 a | 0.02 b | 0 *c | 4.59 ad | 8.09 d | 0.95 ab | 0.47 a |
| Ssd-00003 | 0.04 a | 3.45 b | 12.84 b | 2.98 b | 4.80 b | 3.23 b | 2.78 b |
| Ssd-00007 | 5.56 a | 0.21 b | 0.33 b | <0.01 c | 0.05 c | <0.01 c | 0.02 c |
| Ssd-00014 | 0.04 a | 0.44 b | 1.48 c | 0.11 ad | 0.11 ad | 0.14 d | 0.61 bd |
| Ssd-00019 | 0.49 a | 0.28 a | 1.67 bd | 1.42 a | 0.73 ab | 3.91 c | 1.80 cd |
| Ssd-00039 | <0.01 a | 0.80 b | 1.15 b | 8.07 c | 2.32 b | 28.80 d | 21.25 e |
| Ssd-00042 | 0.09 abcd | 0.03 acd | 0.05 ac | <0.01 b | 1.88 cd | <0.01 d | 0.01 abcd |
| Ssd-00048 | <0.01 a | 0.31 b | 0.44 b | 1.93 c | 0.57 b | 4.56 d | 5.31 d |
| Ssd-00059 | 2.04 a | <0.01 b | <0.01 b | 0.08 c | 0.03 c | 0.06 bc | 0.07 c |
| Ssd-00064 | <0.01 a | 0.32 b | 0.29 b | 2.57 c | 3.62 c | 1.80 c | 2.17 c |
| Ssd-00071 | 0.04 a | 0.05 a | 0.02 a | 0.06 a | 1.48 b | 0.06 a | 1.12 b |
| Ssd-00134 | 0.04 a | 5.17 b | 4.48 b | 4.49 c | 0.50 c | 0.76 c | 1.68 bc |
| Ssd-00188 | <0.01 a | 0.81 bc | 0.94 bd | 0.17 c | 1.87 bd | 2.33 d | 4.21 e |
| Ssd-00331 | 0 *a | 0.02 b | 0.02 b | 0.49 c | 0.26 bc | 1.90 d | 0.89 e |
| Ssd-00928 | 0.21 ad | 2.06 b | 0.55 ac | 1.13 bc | 0.55 ac | 0.10 d | 0.03 d |
| Ssd-00930 | 4.28 a | 1.20 a | 0.39 b | 0.54 ab | 4.82 ab | 0.16 b | 0.14 b |
| Ssd-01090 | 2.54 a | 0.20 b | 0.35 bc | 0.12 b | 0.08 bcd | 0.01 cd | 0.01 d |
| Ssd-01177 | 1.82 a | 0.03 bd | 0.01 b | 0 *c | <0.01 bc | 0 *c | <0.01 cd |
| Ssd-01244 | 3.90 a | 0.23 b | 0.05 c | 0.06 cd | 0.01 d | 0 *e | 0 *e |
| Ssd-01332 | 0 *a | 0.08 b | <0.01 a | 0.15 bc | <0.01 ac | 0.50 d | 1.12 d |
| Ssd-01334 | 2.82 a | 0 *b | 0 *b | <0.01 b | <0.01 b | 0 *b | 0 *b |
| Ssd-01381 | 1.14 a | <0.01 b | 0 *b | 0 *b | <0.01 b | 0 *b | 0 *b |
| OTUs | Closest Valid Taxon | PI * | Taxonomic Affiliation |
|---|---|---|---|
| Ssd-00001 | Lactobacillus amylovorus | 99.7% | (Lactobacillaceae) |
| Ssd-00003 | Prevotella copri | 97.8% | (Prevotellaceae) |
| Ssd-00007 | Escherichia fergusonii | 98.9% | (Enterobacteriaceae) |
| Ssd-00014 | Terrisporobacter mayombei | 97.3% | (Peptostreptococcaceae) |
| Ssd-00019 | Lactobacillus reuteri | 98.6% | (Lactobacillaceae) |
| Ssd-00039 | Streptococcus alactolyticus | 100% | (Streptococcaceae) |
| Ssd-00042 | Lactobacillus salivarius | 99.3% | (Lactobacillaceae) |
| Ssd-00048 | Streptococcus alactolyticus | 96.5% | (Streptococcaceae) |
| Ssd-00059 | Lactobacillus vaginalis | 98.3% | (Lactobacillaceae) |
| Ssd-00064 | Blautia luti | 97.6% | (Lachnospiraceae) |
| Ssd-00071 | Megasphaera elsdenii | 98% | (Veillonellaceae) |
| Ssd-00134 | Clostridium saccharoperbutylacetonicum | 97% | (Clostridiaceae) |
| Ssd-00188 | Eubacterium rectale | 99.2% | (Lachnospiraceae) |
| Ssd-00331 | Streptococcus alactolyticus | 97.5% | (Streptococcaceae) |
| Ssd-00928 | Ruminococcus gnavus | 95.6% | (Lachnospiraceae) |
| Ssd-00930 | Prevotella stercorea | 96% | (Prevotellaceae) |
| Ssd-01090 | Paludicola psychrotolerans | 88.3% | (Ruminococcaceae) |
| Ssd-01177 | Bacteroides vulgatus | 99.8% | (Bacteroidaceae) |
| Ssd-01244 | Ruminococcus bromii | 91.3% | (Ruminococcaceae) |
| Ssd-01332 | Catenibacterium mitsuokai | 94.5% | (Erysipelotrichaceae) |
| Ssd-01334 | Bacteroides fragilis | 98.7% | (Bacteroidaceae) |
| Ssd-01381 | Enterocloster bolteae | 95.7% | (Lachnospiraceae) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fresno Rueda, A.; Samuel, R.; St-Pierre, B. Investigating the Effects of a Phytobiotics-Based Product on the Fecal Bacterial Microbiome of Weaned Pigs. Animals 2021, 11, 1950. https://doi.org/10.3390/ani11071950
Fresno Rueda A, Samuel R, St-Pierre B. Investigating the Effects of a Phytobiotics-Based Product on the Fecal Bacterial Microbiome of Weaned Pigs. Animals. 2021; 11(7):1950. https://doi.org/10.3390/ani11071950
Chicago/Turabian StyleFresno Rueda, Anlly, Ryan Samuel, and Benoit St-Pierre. 2021. "Investigating the Effects of a Phytobiotics-Based Product on the Fecal Bacterial Microbiome of Weaned Pigs" Animals 11, no. 7: 1950. https://doi.org/10.3390/ani11071950
APA StyleFresno Rueda, A., Samuel, R., & St-Pierre, B. (2021). Investigating the Effects of a Phytobiotics-Based Product on the Fecal Bacterial Microbiome of Weaned Pigs. Animals, 11(7), 1950. https://doi.org/10.3390/ani11071950