
Past and Recent Effects of Livestock Activity on the Genetic Diversity and Population Structure of Native Guanaco Populations of Arid Patagonia

 and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Microsatellite Analysis
2.3. Mitochondrial DNA
2.4. Estimation and Delimitation of Genetic Units and Gene Flow
2.5. Demographic History
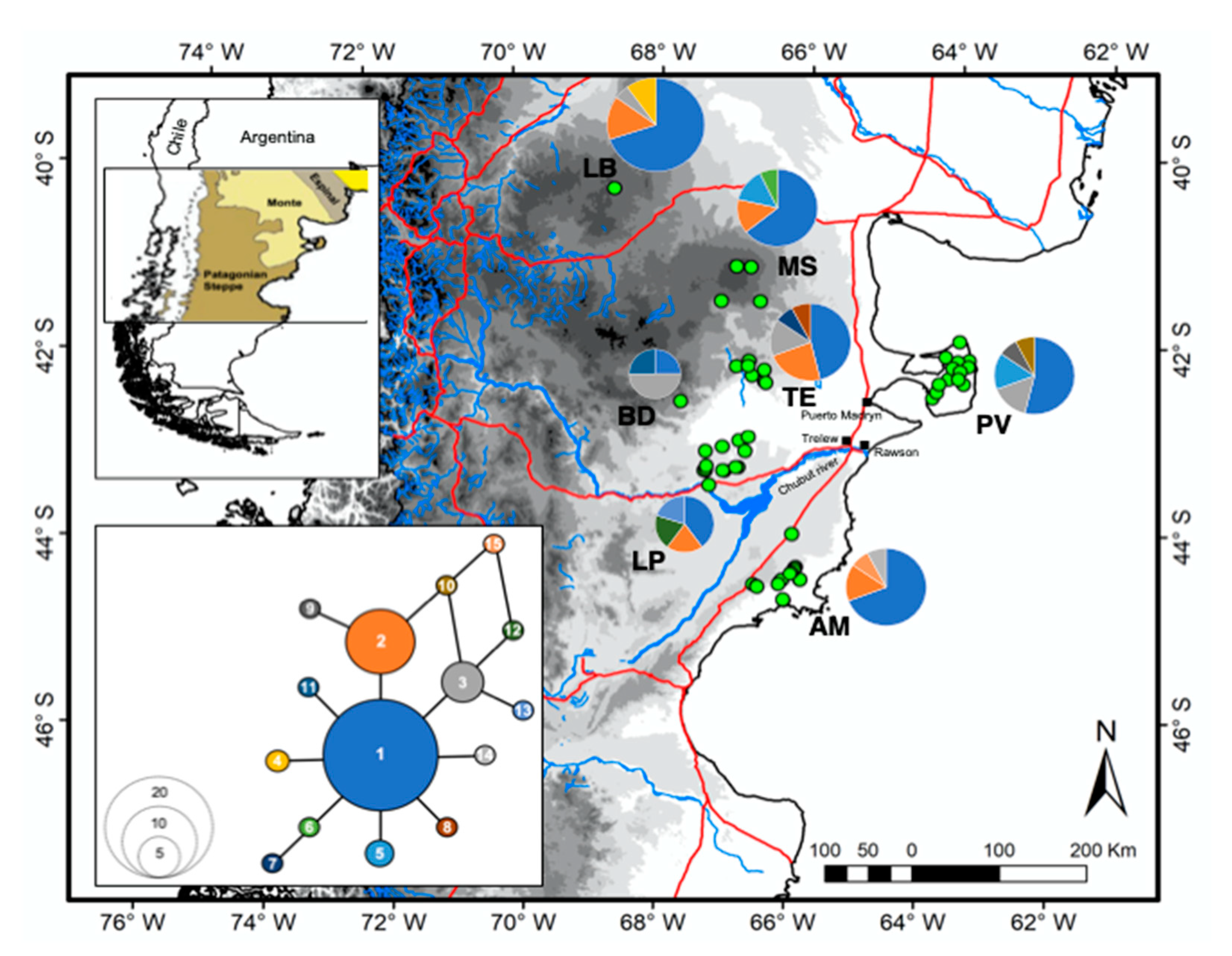
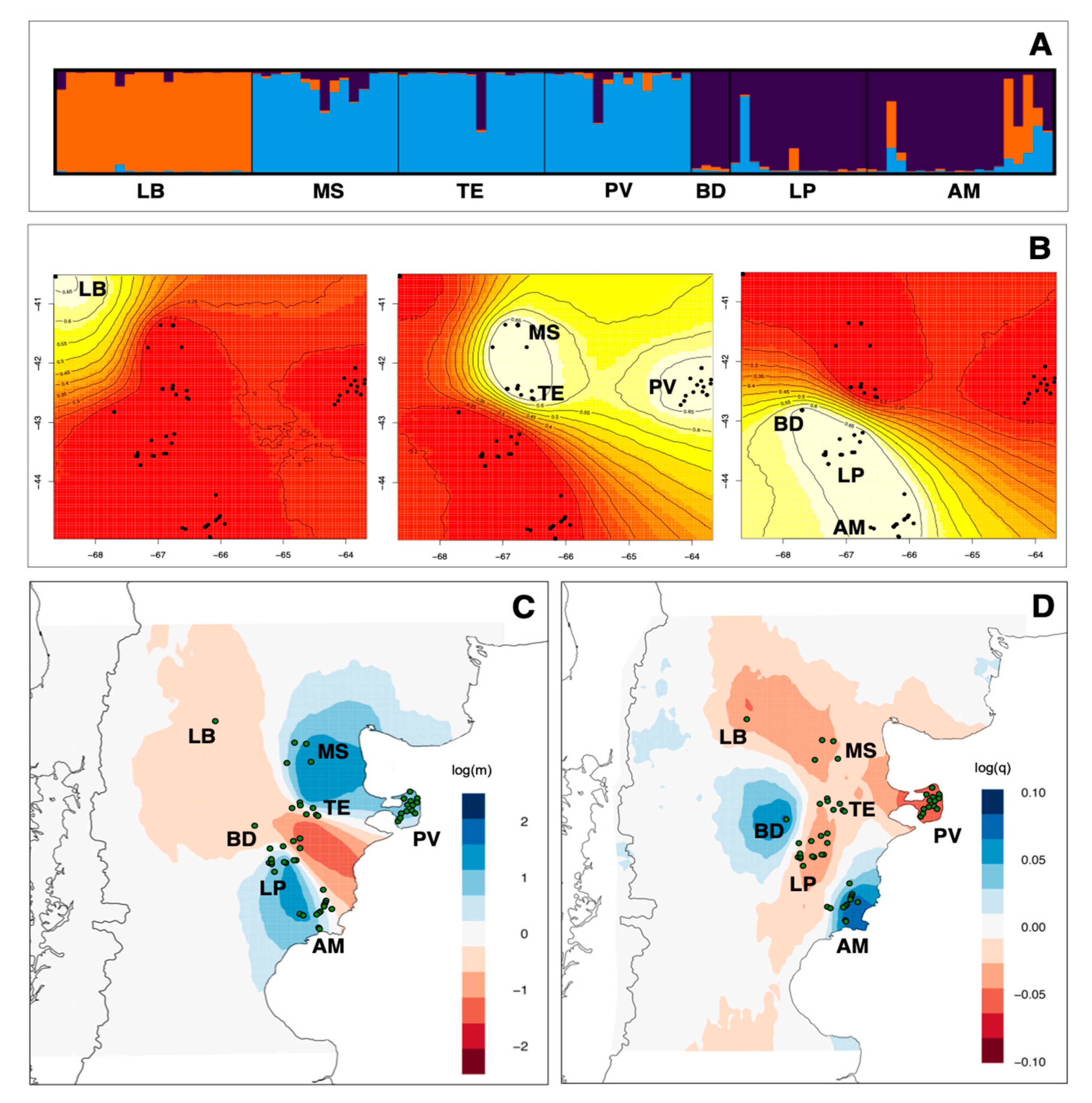
3. Results
3.1. Genetic Diversity
3.2. Genetic Structure and Gene Flow
3.3. Demographic History
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sbordoni, V.; Allegrucci, G.; Cesaroni, D. Population Structure. In Encyclopedia of Caves; White, W.B., Culver, D.C., Eds.; Academic Press: Chennai, India, 2012; pp. 608–618. [Google Scholar]
- Yannic, G.; Ortego, J.; Pellissier, L.; Lecomte, N.; Bernatchez, L.; Côté, S.D. Linking genetic and ecological differentiation in an ungulate with a circumpolar distribution. Ecography 2018, 41, 922–937. [Google Scholar] [CrossRef]
- Epps, C.W.; Palsbøll, P.J.; Wehausen, J.D.; Roderick, G.K.; Ramey, R.R.; McCullough, D.R. Highways block gene flow and cause a rapid decline in genetic diversity of desert bighorn sheep. Ecol. Lett. 2005, 8, 1029–1038. [Google Scholar] [CrossRef]
- Ito, T.Y.; Lhagvasuren, B.; Tsunekawa, A.; Shinoda, M.; Takatsuki, S.; Buuveibaatar, B.; Chimeddorj, B. Fragmentation of the Habitat of Wild Ungulates by Anthropogenic Barriers in Mongolia. PLoS ONE 2013, 8, e56995. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.L.; Ironside, K.; Wayne, R.K.; Boydston, E.E. Connectivity of mule deer (Odocoileus hemionus) populations in a highly fragmented urban landscape. Landsc. Ecol. 2019, 34, 1097–1115. [Google Scholar] [CrossRef]
- de Jager, D.; Harper, C.K.; Bloomer, P. Genetic diversity, relatedness and inbreeding of ranched and fragmented Cape buffalo population in southern Africa. PLoS ONE 2020, 15, e0236717. [Google Scholar] [CrossRef] [PubMed]
- Pierson, J.C.; Coates, D.J.; Oostermeijer, J.G.B.; Beissinger, S.R.; Bragg, J.G.; Sunnucks, P.; Schumaker, N.H.; Young, A.G. Genetic factors in threatened species recovery plans on three continents. Front. Ecol. Env. 2016, 14, 433–440. [Google Scholar] [CrossRef]
- Ross, S.; Costanzi, J.-M.; Jahdhami, M.A.; Rawahi, H.A.; Ghazali, M.; Senn, H. First evaluation of the population structure, genetic diversity and landscape connectivity of the Endangered Arabian tahr. Mamm. Biol. 2020, 100, 659–673. [Google Scholar] [CrossRef]
- León, R.J.C.; Bran, D.; Collantes, M.; Paruelo, J.M.; Soriano, A. Grandes unidades de vegetación de la Patagonia extra andina. Ecol. Austral 1998, 8, 125–144. [Google Scholar]
- González, B.A.; Palma, R.E.; Zapata, B.; Marín, J.C. Taxonomic and biogeographical status of guanaco Lama guanicoe (Artiodactyla, Camelidae). Mammal Rev. 2006, 36, 157–178. [Google Scholar] [CrossRef]
- Hoffstetter, R. High Andean mammalian faunas during th Oxford e Plio-Pleistocene. In High Altitude Tropical Bioge-Ography; Vulleumier, F., Monasterio, M., Eds.; Oxford University Press: Oxford, UK, 1986; pp. 218–245. [Google Scholar]
- MacQuarrie, N.; Horton, B.K.; Zandt, J.; Beck, S.; Decelles, P.G. Lithospheric evolution of the Andean fold–thrust belt Bolivia and the origin of the central Andean Plateau. Ectonophysics 2005, 399, 15–37. [Google Scholar] [CrossRef]
- Rabassa, J.; Coronato, A.; Martinez, O. Late Cenozoic glaciations in Patagonia and Tierra del Fuego: An updated review. Biol. J. Linn. Soc. 2011, 103, 316–335. [Google Scholar] [CrossRef]
- Marín, J.C.; González, B.A.; Poulin, E.; Casey, C.; Johnson, W.E. The influence of the arid Andean high plateau on the phylogeography and population genetics of guanaco (Lama guanicoe) in South America. Mol. Ecol. 2013, 22, 463–482. [Google Scholar] [CrossRef]
- Baldi, R.B.; Acebes, P.; Cuéllar, E.; Funes, M.; Hoces, D.; Puig, S.; Franklin, W.L. Lama guanicoe. IUCN Red List Threat. Species 2016, e.T11186A18540211. [Google Scholar] [CrossRef]
- Franklin, W.L.; Bas, F.; Bonacic, C.F.; Cunazza, C.; Soto, N. Striving to manage Patagonian guanacos for sustained use in the grazing agroecosystems of southern Chile. Wildl. Soc. Bull. 1997, 25, 65–73. [Google Scholar]
- Cunazza, C.; Puig, S.; Villalba, L. Situación actual del guanaco y su ambiente. In Técnicas para el Manejo del Guanaco; Puig, G.S., Ed.; Grupo Especialista en Camélidos Sudamericanos, Comisión de Supervivencia de Especies, UICN: Phnom Penh, Cambodia, 1995; Chapter 2; pp. 27–50. [Google Scholar]
- Soriano, A.; Movia, C.P. Erosión y desertización en la Patagonia. Interciencia 1986, 11, 77–83. [Google Scholar]
- Baldi, R.; Albon, S.D.; Elston, D. Guanacos and sheep: Evidence for continuing competition in arid Patagonia. Oecologia 2001, 129, 561–570. [Google Scholar] [CrossRef]
- Baldi, R.; Pelliza-Sbriller, A.; Elston, D.; Albon, S. High potential for competition between guanacos and sheep in Patagonia. J. Wildl. Manag. 2004, 68, 924–938. [Google Scholar] [CrossRef]
- González, B.A.; Orozco-terWengel, P.; von Borries, R.; Johnson, W.E.; Franklin, W.L.; Marín, J.C. Maintenance of Genetic Diversity in an Introduced Island Population of Guanacos after Seven Decades and Two Severe Demographic Bottlenecks: Implications for Camelid Conservation. PLoS ONE 2014, 9, e91714. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritschi, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Lang, K.; Wang, Y.; Plante, Y. Fifteen polymorphic dinucleotide microsatellites in llamas and alpacas. Anim. Genet. 1996, 27, 293. [Google Scholar] [CrossRef]
- Penedo, M.C.T.; Casetano, A.R.; Cordova, K.I. Microsatellite markers for South American camelids. Anim. Genet. 1998, 29, 411–412. [Google Scholar]
- Penedo, M.; Caetano, A.; Cordova, K. Six microsatellite markers for South American camelids. Anim. Genet. 1999, 30, 382–405. [Google Scholar] [CrossRef]
- Sarno, R.J.; David, V.A.; Franklin, W.L.; O’Brien, S.J.; Johnson, W.E. Development of microsatellite markers in the guanaco, Lama guanicoe: Utility for South American camelids. Mol. Ecol. 2000, 9, 1922–1924. [Google Scholar] [CrossRef]
- Taberlet, P.; Griffin, S.; Goossens, B.; Questiau, S.; Manceau, V.; Escaravage, N.; Waits, L.P.; Bouvet, J. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996, 24, 3189–3194. [Google Scholar] [CrossRef] [PubMed]
- Park, S.D.E. Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. Ph.D. Thesis, University College Dublin, Dublin, Ireland, 2001. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.; Cockerham, C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (Version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Marín, J.; Casey, C.; Kadwell, M.; Yaya, K.; Hoces, D.; Olazabal, J.; Rosadio, R.; Rodriguez, J.; Spotorno, A.; Bruford, M.W.; et al. Mitochondrial phylogeography and demographic history of the Vicuña: Implications for conservation. Heredity 2007, 99, 70–80. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1655–1659. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Guillot, G.; Mortier, F.; Estoup, A. Geneland: A computer package for landscape genetics. Mol. Ecol. Notes 2005, 5, 712–715. [Google Scholar] [CrossRef]
- Petkova, D.; Novembre, J.; Stephens, M. Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 2016, 48, 94–100. [Google Scholar] [CrossRef]
- Kimura, M.; Weiss, G.H. The Stepping Stone Model of Population Structure and the Decrease of Genetic Correlation with Distance. Genetics 1964, 49, 561–576. [Google Scholar] [CrossRef]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar]
- Miller, M.P. Alleles in Space (AIS): Computer Software for the Joint Analysis of Interindividual Spatial and Genetic Information. J. Hered. 2005, 96, 722–724. [Google Scholar] [CrossRef]
- Storz, J.F.; Beaumont, M.A. Testing for genetic evidence of population expansion and contraction: An empirical analysis of microsatellite dna variation using a hierarchical bayesian model. Evolution 2002, 56, 154–166. [Google Scholar] [CrossRef]
- Weber, J.L.; Wong, C. Mutation of human short tandem repeats. Hum. Mol. Genet. 1993, 2, 1123–1128. [Google Scholar] [CrossRef]
- Di Rienzo, A.; Peterson, A.C.; Garza, J.C.; Valdes, A.M.; Slatkin, M.; Freimer, N.B. Mutational processes of simple sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 1994, 91, 3166–3170. [Google Scholar] [CrossRef]
- Brohede, J.; Primmer, C.R.; Møller, A.; Ellegren, H. Heterogeneity in the rate and pattern of germline mutation at individual microsatellite loci. Nucleic Acids Res. 2002, 30, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Bulut, Z.; McCormick, C.R.; Gopurenko, D.; Williams, R.N.; Bos, D.H.; DeWoody, J.A. Microsatellite mutation rates in the eastern tiger salamander (Ambystoma tigrinum tigrinum) differ 10-fold across loci. Genetica 2009, 136, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Franklin, W.L. Contrasting socioecologies of South America’s wild camelids: The vicuna and the guanaco. In Advances in the Study of Mammalian Behavior; Eisenberg, J.K., Kleiman, D.G., Eds.; American Society of Mammalogists: Shippenburgs, PA, USA, 1983; pp. 573–629. [Google Scholar]
- Plummer, M.; Best, N.; Cowles, K.; Vines, K. CODA: Convergence diagnosis and output analysis for MCMC. R News 2006, 6, 7–11. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2011. [Google Scholar]
- Greshko, M. Colossal Volcano behind ‘Mystery’ Global Cooling Finally Found. National Geographic. 2019. Available online: https://www.nationalgeographic.com/science/article/colossal-volcano-behind-mystery-global-cooling-found (accessed on 1 March 2021).
- Smith, V.C.; Costa, A.; Aguirre-Diaz, G.; Pedrazzi, D.; Scifo, A.; Plunkett, G.; Poret, M.; Tournigand, P.-Y.; Miles, D.; Dee, M.W.; et al. The magnitude and impact of the 431 CE Tierra Blanca Joven eruption of llopango, El Salvador. Proc. Natl. Acad. Sci. USA 2020, 117, 26061–26068. [Google Scholar] [CrossRef] [PubMed]
- McKee, C.O.; Baille, M.G.; Reimer, P.J. A revised age of AD 667–699 for the latest major eruption at Rabul. Bull. Volcanol. 2015, 77, 65. [Google Scholar] [CrossRef]
- Abbona, C.C.; Neme, G.; Johnson, J.; Gil, A.; Villalba, R.; Nagaoka, L.; Kim, T.; Wolverton, S. Sustainable harvest or resource depression? Using ancient DNA to study the population dynamics of guanaco in western Argentina during the Holocene. J. Archaeol. Sci. 2021, 129, 105355. [Google Scholar] [CrossRef]
- Sarno, R.J.; Jennings, D.E.; Franklin, W.L. Estimating effective population size of guanacos in Patagonia: An integrative approach for wildlife conservation. Conserv. Genet. 2015, 16, 1167–1180. [Google Scholar] [CrossRef]
- Sarno, R.J.; Bank, M.S.; Stern, H.S.; Franklin, W.L. Forced dispersal of juvenile guanacos (Lama guanicoe): Causes, variation, and fates of individuals dispersing at different times. Behav. Ecol. Sociobiol. 2003, 54, 22–29. [Google Scholar] [CrossRef]
- Safner, T.; Buzan, E.; Rezic, A.; Šprem, N. Small-scale spatial genetic structure of Alpine chamois (Rupicapra rupicapra) in Northern Dinarides. Eur. J. Wildl. Res. 2019, 65, 23. [Google Scholar] [CrossRef]
- Edelhoff, H.; Zachos, F.E.; Fickel, J.; Epps, C.W.; Balkenhol, N. Genetic analysis of red deer (Cervus elaphus) administrative management units in a human-dominated landscape. Conserv. Genet. 2020, 21, 261–276. [Google Scholar] [CrossRef]
- Aagesen, D. Crisis and conservation at the end of the world: Sheep ranching in Argentine Patagonia. Environ. Conserv. 2000, 27, 208–215. [Google Scholar] [CrossRef]
- Soriano, A.; and Paruelo, J.M. El pastoreo ovino: Principios ecológicos para el manejo de los campos. Cienc. Hoy. 1990, 2, 44–53. [Google Scholar]
- Pedrana, J.; Bustamante, J.; Travaini, A.; Rodríguez, A. Factors influencing guanaco distribution in southern Argentine Patagonia and implications for its sustainable use. Biodiv. Conserv. 2010, 19, 3499–3512. [Google Scholar] [CrossRef]
- Travaini, A.; Zapata, S.C.; Bustamante, J.; Pedrana, J.; Zanón, J.I.; Rodríguez, A. Guanaco abundance and monitoring in Southern Patagonia: Distance sampling reveals substantially greater numbers than previously reported. Zool. Stud. 2015, 54, 23. [Google Scholar] [CrossRef] [PubMed]
- Antún, M.; Baldi, R. Choosing what is left: The spatial structure of a wild herbivore population within a livestock-dominated landscape. PeerJ 2020, 8, e8945. [Google Scholar] [CrossRef] [PubMed]
- González, B.A. (Facultad de Ciencias Forestales y de la Conservación de la Naturaleza, Universidad de Chile, Santiago 8330015, Chile). Personal communication, 2020.
- Jones, P.F.; Jakes, A.F.; Telander, A.C.; Sawyer, H.; Martin, B.H.; Hebblewhite, M. Fences reduce habitat for a partially migratory ungulate in the Northern Sagebrush Steppe. Ecosphere 2019, 10, e02782. [Google Scholar] [CrossRef]
- de Jong, J.F.; van Hooft, P.; Megens, H.-J.; Crooijmans, R.P.M.A.; de Groot, G.A.; Pemberton, J.M.; Huisman, J.; Bartoš, L.; Iacolina, L.; van Wieren, S.E.; et al. Fragmentation and Translocation Distort the Genetic Landscape of Ungulates: Red Deer in the Netherlands. Front. Ecol. Evol. 2020, 8, 535715. [Google Scholar] [CrossRef]
- Coronato, F.; Pessacg, N.; Alvarez, M.P. The climate of Península Valdés within a regional frame. In Late Cenozoic of Península Valdés, Patagonia, Argentina; Bouza, P., Bilmes, A., Eds.; Springer: Cham, Switzerland, 2017; pp. 85–104. [Google Scholar]
- Novaro, A.J.; Baldi, R.; and Antún, M. Silvestría en Patagonia: ¿qué especies, umbrales y procesos la definen? In Abstracts of Jornadas Argentinas de Mastozoología XXXII; SAREM: Puerto Madryn, Argentina, 2019; p. 61. Available online: http://jam.sarem.org.ar/libro-de-resumenes (accessed on 10 December 2019).
- Marino, A.; Rodríguez, V.; Pazos, G. Resource-defense polygyny and self-limitation of population density in free-ranging guanacos. Behav. Ecol. 2016, 27, 757–765. [Google Scholar] [CrossRef]
- Moraga, C.; Funes, M.; Pizarro, J.; Briceño, C.; Novaro, A. Effects of livestock on guanaco Lama guanicoe density, movements and habitat selection in a forest–grassland mosaic in Tierra del Fuego, Chile. Oryx 2015, 49, 30–41. [Google Scholar] [CrossRef]
- Mueller, T.; Olson, K.A.; Dressler, G.; Leimgruber, P.; Fuller, T.K.; Nicolson, C.; Novaro, A.J.; Bolgeri, M.J.; Wattles, D.; DeStefano, S.; et al. How landscape dynamics link individual- to population-level movement patterns: A multispecies comparison of ungulate relocation data. Glob. Ecol. Biogeogr. 2011, 20, 683–694. [Google Scholar] [CrossRef]
- Wildlife Friendly Enterprise Network, 2015. Available online: http://wildlifefriendly.org/wfen-welcomes-wildlifeconservation-society-argentina-first-certified-wool-producers-from-iconic-peninsula-valdes/ (accessed on 12 March 2020).



{kind=link}
{kind=link}
| Localities | mtDNA | Microsatellites | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | n | np | h ± SD | p ± SD | N | A ± SD | PA | AR ± SD | H0 ± SD | HE ± SD | FIS ± SD | |
| Group 1: Loma Blanca | 20 | 4 | 1 | 0.5 ± 0.12 | 0.002 ± 0.001 | 20 | 7.533 ± 3.18 | 20 | 1.762 ± 0.10 | 0.905 ± 0.09 | 0.762 ± 0.1 | −0.208 ± 0.17 |
| Group 2 | 45 | 8.933 ± 4.86 | 7.242 ± 3.27 | −0.028 ± 0.26 | ||||||||
| Meseta Somuncura | 14 | 4 | 1 | 0.582 ± 0.14 | 0.002 ± 0.001 | 15 | 6.867 ± 3.66 | 3 | 1.726 ± 0.22 | 0.778 ± 0.26 | 0.726 ± 0.22 | −0.082 ± 0.23 |
| Telsen | 13 | o | 2 | 0.756 ± 0.1 | 0.004 ± 0.001 | 15 | 7.067 ± 2.96 | 3 | 1.708 ± 0.13 | 0.756 ± 0.19 | 0.764 ± 0.12 | 0.017 ± 0.21 |
| Península Valdés | 13 | 5 | 2 | 0.705 ± 0.12 | 0.004 ± 0.001 | 15 | 6.267 ± 2.96 | 4 | 1.764 ± 0.12 | 0.783 ± 0.25 | 0.708 ± 0.13 | −0.094 ± 0.31 |
| Group 3 | 37 | 8.866 ± 3.4 | 7.512 ± 2.51 | 0.152 ± 0.22 | ||||||||
| Bajada del Diablo | 4 | 3 | 1 | 0.833 ± 0.22 | 0.004 ± 0.001 | 4 | 3.667 ± 1.11 | 2 | 1.722 ± 0.15 | 0.617 ± 0.29 | 0.722 ± 0.15 | 0.128 ± 0.41 |
| Las Plumas | 5 | 4 | 2 | 0.900 ± 0.16 | 0.006 ± 0.001 | 14 | 5.933 ± 2.28 | 4 | 1.720 ± 0.14 | 0.673 ± 0.27 | 0.720 ± 0.14 | 0.099 ± 0.30 |
| Ameghino | 13 | 4 | 2 | 0.526 ± 0.15 | 0.003 ± 0.001 | 19 | 7.333 ± 2.44 | 13 | 1.762 ± 0.09 | 0.635 ± 0.19 | 0.762 ± 0.09 | 0.165 ± 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesas, A.; Baldi, R.; González, B.A.; Burgi, V.; Chávez, A.; Johnson, W.E.; Marín, J.C. Past and Recent Effects of Livestock Activity on the Genetic Diversity and Population Structure of Native Guanaco Populations of Arid Patagonia. Animals 2021, 11, 1218. https://doi.org/10.3390/ani11051218
Mesas A, Baldi R, González BA, Burgi V, Chávez A, Johnson WE, Marín JC. Past and Recent Effects of Livestock Activity on the Genetic Diversity and Population Structure of Native Guanaco Populations of Arid Patagonia. Animals. 2021; 11(5):1218. https://doi.org/10.3390/ani11051218
Chicago/Turabian StyleMesas, Andrés, Ricardo Baldi, Benito A. González, Virginia Burgi, Alexandra Chávez, Warren E. Johnson, and Juan C. Marín. 2021. "Past and Recent Effects of Livestock Activity on the Genetic Diversity and Population Structure of Native Guanaco Populations of Arid Patagonia" Animals 11, no. 5: 1218. https://doi.org/10.3390/ani11051218
APA StyleMesas, A., Baldi, R., González, B. A., Burgi, V., Chávez, A., Johnson, W. E., & Marín, J. C. (2021). Past and Recent Effects of Livestock Activity on the Genetic Diversity and Population Structure of Native Guanaco Populations of Arid Patagonia. Animals, 11(5), 1218. https://doi.org/10.3390/ani11051218