
Dynamic Changes in Fecal Microbial Communities of Neonatal Dairy Calves by Aging and Diarrhea

 , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Diet
2.2. Monitoring of Animal Health
2.3. Feces Collection
2.4. DNA Purification
2.5. 16S rRNA Gene Sequencing and Sequence Analysis
2.6. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) and Linear Discriminant Analysis Effect Size (LEfSe) Analysis
2.7. Statistical Analysis
3. Results
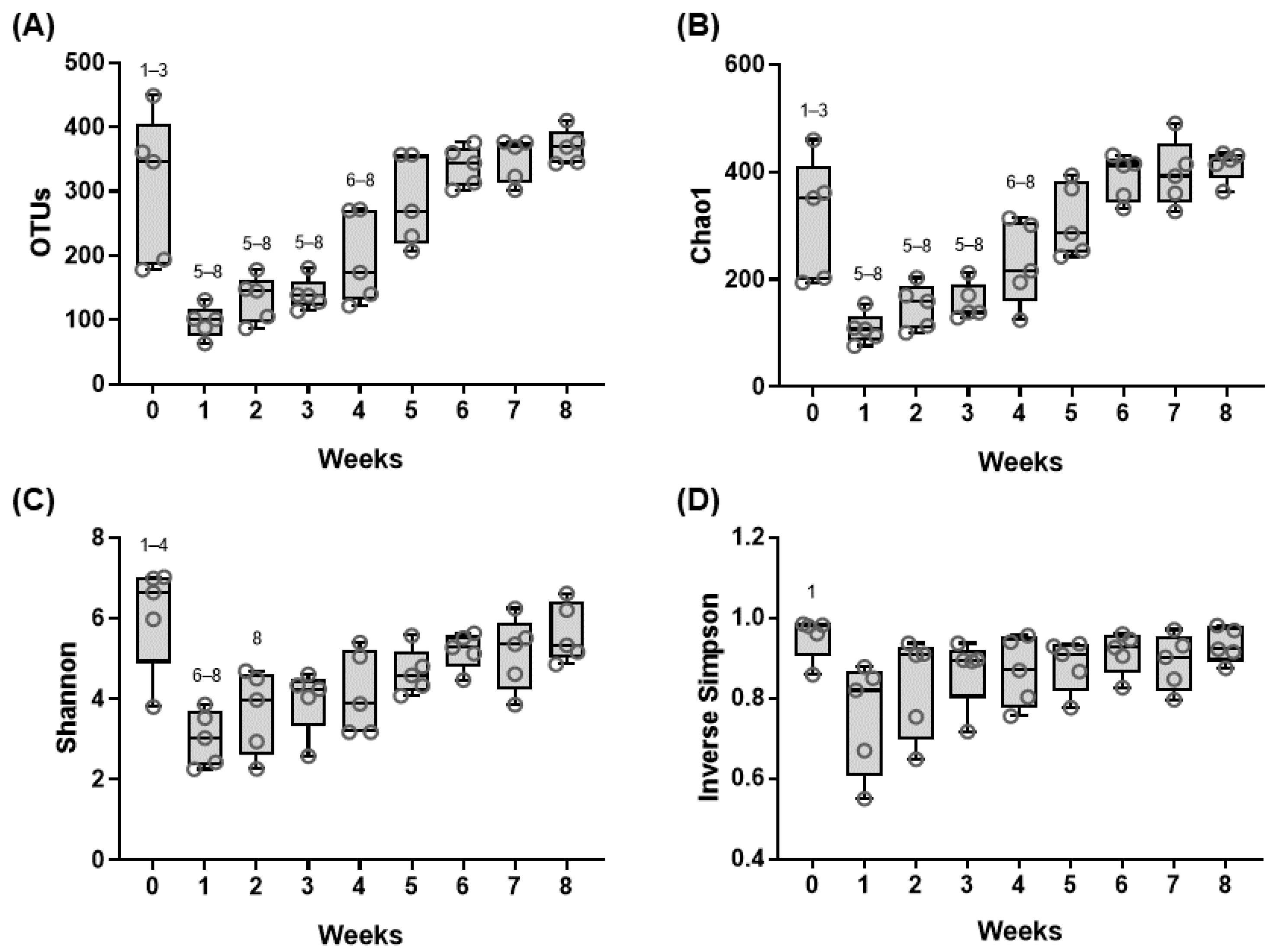
3.1. Sequencing Information
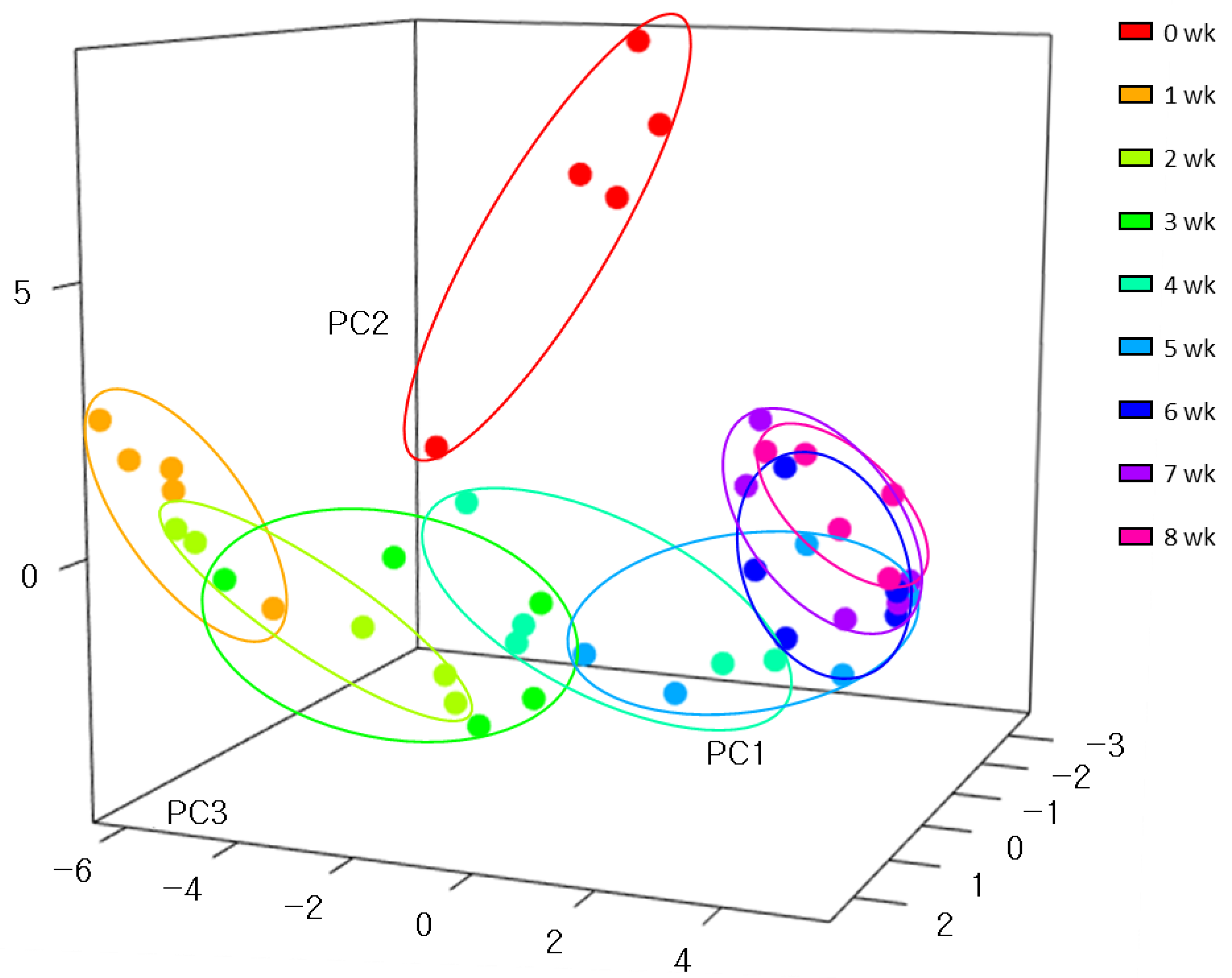
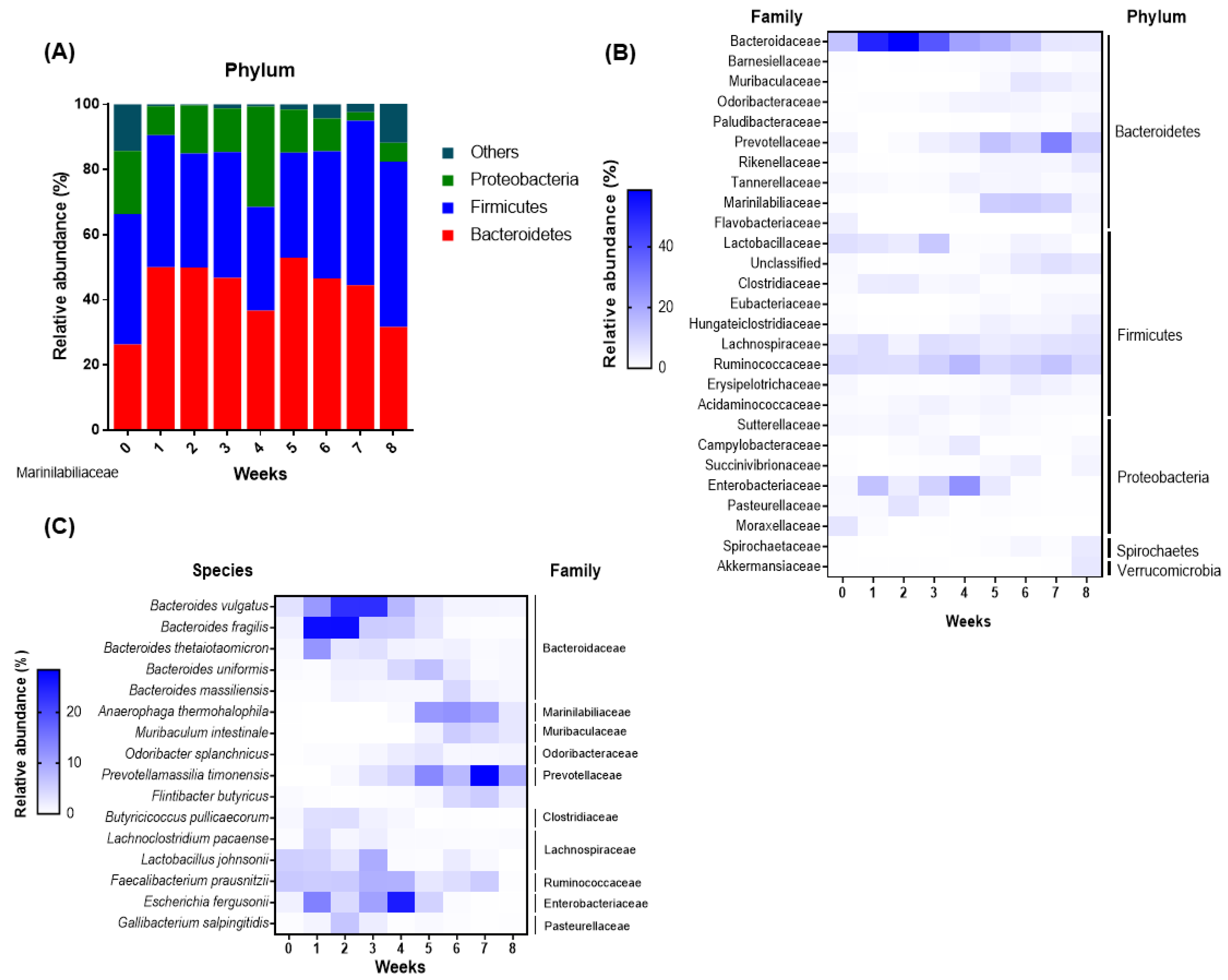
3.2. Shifts in the Fecal Microbiome Composition of Dairy Calves during the Pre-Weaning Period
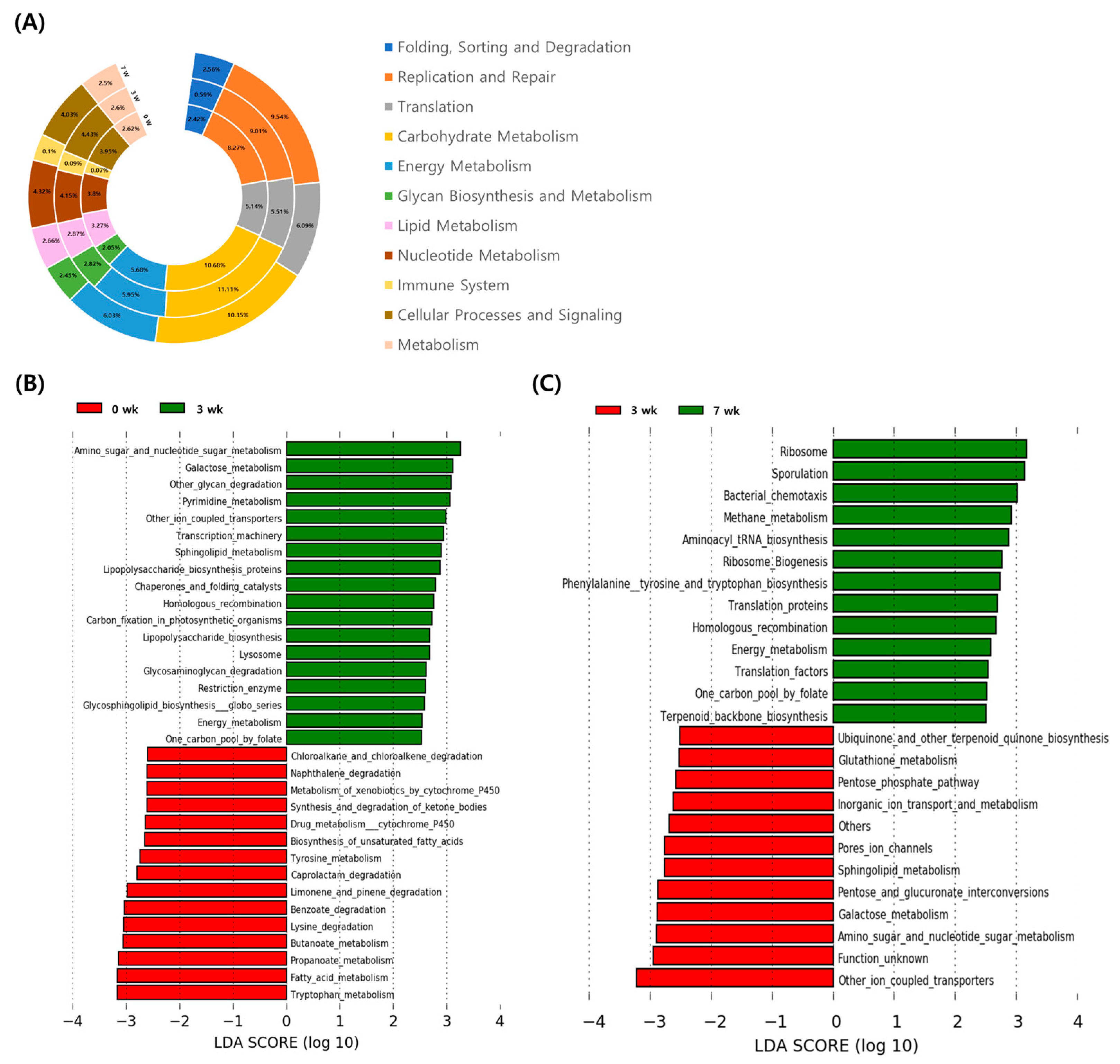
3.3. Changes of Functional Gene Family in Fecal Microbiome during the Pre-Weaning Period
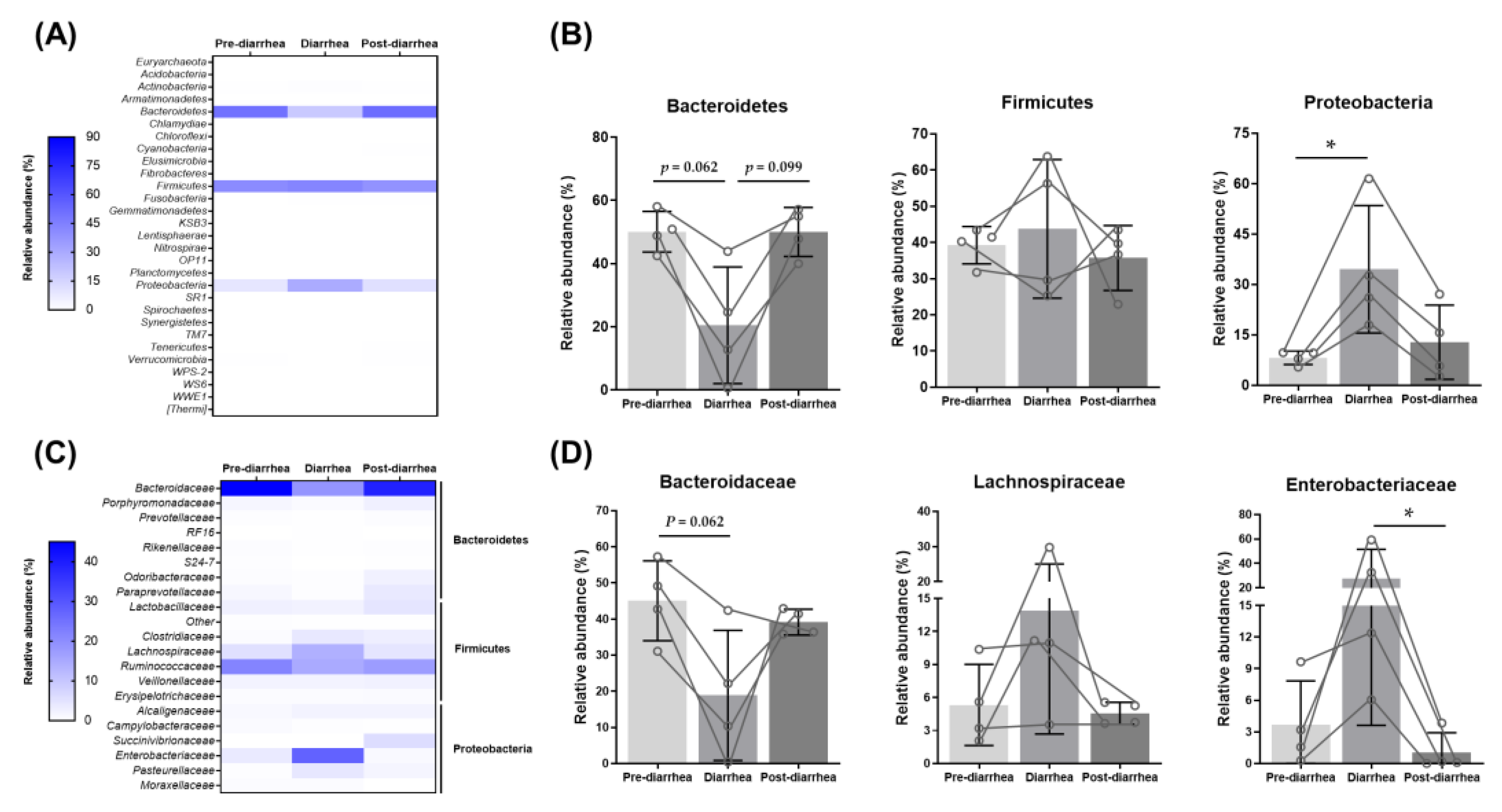
3.4. Shift in the Fecal Microbiome during Diarrheal Disease
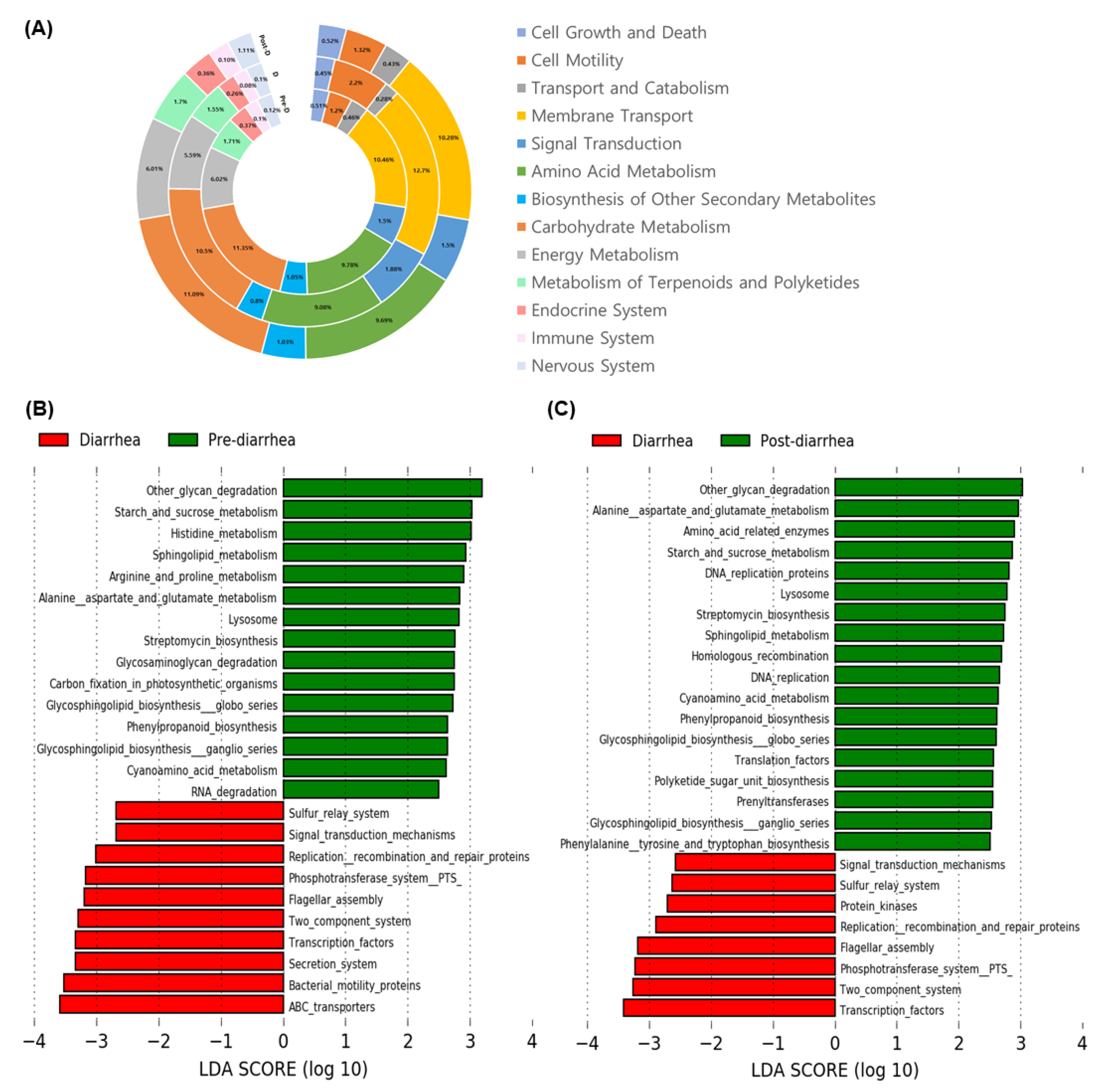
3.5. Changes in Abundance of Functional Microbial Genes Family by Diarrhea
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warner, R.G.; Flatt, W.P.; Loosli, J.K. Dietary factors influencing the development of the ruminant stomach. J. Agric. Food Chem. 1956, 4, 788–792. [Google Scholar] [CrossRef]
- Bauer, E.; Williams, B.A.; Smidt, H.; Verstegen, M.W.A.; Mosenthin, R. Influence of the gastrointestinal microbiota on devel-opment of the immune system in young animals. Curr. Issues Intest. Microbiol. 2006, 7, 35–51. [Google Scholar] [PubMed]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; et al. Gut Immune Maturation Depends on Colonization with a Host-Specific Microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.; McLeod, K.; Klotz, J.; Heitmann, R. Rumen Development, Intestinal Growth and Hepatic Metabolism In The Pre- and Postweaning Ruminant. J. Dairy Sci. 2004, 87, E55–E65. [Google Scholar] [CrossRef]
- Carberry, C.A.; Kenny, D.A.; Han, S.; McCabe, M.S.; Waters, S.M. Effect of Phenotypic Residual Feed Intake and Dietary Forage Content on the Rumen Microbial Community of Beef Cattle. Appl. Environ. Microbiol. 2012, 78, 4949–4958. [Google Scholar] [CrossRef]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed effi-ciency. PLoS ONE. 2014, 9, e85423. [Google Scholar]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal Bacterial Community Composition in Dairy Cows Is Dynamic over the Course of Two Lactations and Correlates with Feed Efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.; Enjalbert, F.; Combes, S.; Cauquil, L.; Bouchez, O.; Monteils, V. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 2014, 116, 245–257. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef]
- Uyeno, Y.; Sekiguchi, Y.; Kamagata, Y. rRNA-based analysis to monitor succession of faecal bacterial communities in Holstein calves. Lett. Appl. Microbiol. 2010, 51, 570–577. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Guan, L.L. Gut microbiome and omics: A new definition to ruminant production and health. Anim. Front. 2016, 6, 8–12. [Google Scholar] [CrossRef]
- Cho, Y.; Yoon, K.J. An overview of calf diarrhea-infectious etiology, diagnosis, and intervention. J. Vet. Sci. 2014, 15, 1–17. [Google Scholar] [CrossRef]
- Heinrichs, A.; Heinrichs, B. A prospective study of calf factors affecting first-lactation and lifetime milk production and age of cows when removed from the herd. J. Dairy Sci. 2011, 94, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-I.; Han, J.-I.; Wang, C.; Cooper, V.; Schwartz, K.; Engelken, T.; Yoon, K.-J. Case–control study of microbiological etiology associated with calf diarrhea. Vet. Microbiol. 2013, 166, 375–385. [Google Scholar] [CrossRef]
- Trinh, P.; Zaneveld, J.R.; Safranek, S.; Rabinowitz, P.M. One Health Relationships Between Human, Animal, and Environmental Microbiomes: A Mini-Review. Front. Public Health 2018, 6, 235. [Google Scholar] [CrossRef]
- Esser, D.; Lange, J.; Marinos, G.; Sieber, M.; Best, L.; Prasse, D.; Bathia, J.; Rühlemann, M.C.; Boersch, K.; Jaspers, C.; et al. Functions of the Microbiota for the Physiology of Animal Metaorganisms. J. Innate Immun. 2019, 11, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-J.; Hase, K. Gut microbiota–generated metabolites in animal health and disease. Nat. Chem. Biol. 2014, 10, 416–424. [Google Scholar] [CrossRef]
- Park, W. Gut microbiomes and their metabolites shape human and animal health. J. Microbiol. 2018, 56, 151–153. [Google Scholar] [CrossRef]
- Song, S.J.; Woodhams, D.C.; Martino, C.; Allaband, C.; Mu, A.; Javorschi-Miller-Montgomery, S.; Suchodolski, J.S.; Knight, R. Engineering the microbiome for animal health and conservation. Exp. Biol. Med. 2019, 244, 494–504. [Google Scholar] [CrossRef]
- Gerber, G.K. The dynamic microbiome. FEBS Lett. 2014, 588, 4131–4139. [Google Scholar] [CrossRef]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef]
- Pilla, R.; Suchodolski, J.S. The Role of the Canine Gut Microbiome and Metabolome in Health and Gastrointestinal Disease. Front. Vet. Sci. 2020, 6, 498. [Google Scholar] [CrossRef] [PubMed]
- Mondo, E.; Marliani, G.; Accorsi, P.A.; Cocchi, M.; Di Leone, A. Role of gut microbiota in dog and cat’s health and diseases. Open Vet. J. 2019, 9, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Hills, J.R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Larson, L.; Owen, F.; Albright, J.; Appleman, R.; Lamb, R.; Muller, L. Guidelines Toward More Uniformity in Measuring and Reporting Calf Experimental Data. J. Dairy Sci. 1977, 60, 989–991. [Google Scholar] [CrossRef]
- Magoč, T.; Magoč, M.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef]
- Hill, M.O. Diversity and Evenness: A Unifying Notation and Its Consequences. Ecology 1973, 54, 427–432. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric formicrobial com-munity comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- DeSantis, T.Z.D., Jr.; Hugenholtz, P.; Keller, K.; Brodie, E.L.; Larsen, N.; Piceno, Y.M.; Phan, R.; Andersen, G.L. NAST: A multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006, 34, W394–W399. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein anno-tation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.; Marcondes, M.I.; De Souza, S.M.; Silva, B.C.D.M.E.; Noronha, M.F.; Resende, R.T.; Machado, F.S.; Mantovani, H.C.; Dill-McFarland, K.A.; Suen, G. Bacterial Community Dynamics across the Gastrointestinal Tracts of Dairy Calves during Preweaning Development. Appl. Environ. Microbiol. 2018, 84, e02675-17. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef]
- Klein-Jöbstl, D.; Quijada, N.M.; Dzieciol, M.; Feldbacher, B.; Wagner, M.; Drillich, M.; Schmitz-Esser, S.; Mann, E. Microbiota of newborn calves and their mothers reveals possible transfer routes for newborn calves’ gastrointestinal microbiota. PLoS ONE 2019, 14, e0220554. [Google Scholar] [CrossRef] [PubMed]
- Malmuthuge, N.; Griebel, P.J.; Guan, L.L. The Gut Microbiome and Its Potential Role in the Development and Function of Newborn Calf Gastrointestinal Tract. Front. Vet. Sci. 2015, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Malmuthuge, N.; Liang, G.; Guan, L.L. Regulation of rumen development in neonatal ruminants through microbial meta-genomes and host transcriptomes. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin, V.; Ransom, L.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Govil, K.; Yadav, D.S.; Patil, A.K.; Nayak, S.; Baghel, R.P.S.; Yadav, P.K.; Malapure, C.D.; Thakur, D. Feeding management for early rumen development in calves. J. Entomol. Zool. Stud. 2017, 5, 1132–1139. [Google Scholar]
- Meale, S.J.; Li, S.C.; Azevedo, P.; Derakhshani, H.; Devries, T.J.; Plaizier, J.C.; Steele, M.A.; Khafipour, E. Weaning age influences the severity of gastrointestinal microbiome shifts in dairy calves. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Zhang, T.; Si, H.; Nan, W.; Xu, C.; Guan, L.; Wright, A.-D.G.; Li, G. The Development of Microbiota and Metabolome in Small Intestine of Sika Deer (Cervus nippon) from Birth to Weaning. Front. Microbiol. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Ruiz, D.R.; Eabecia, L.; Newbold, C.J. Manipulating rumen microbiome and fermentation through interventions during early life: A review. Front. Microbiol. 2015, 6, 1133. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Malmuthuge, N.; Li, F.; Guan, L.L. Colostrum feeding shapes the hindgut microbiota of dairy calves during the first 12 h of life. FEMS Microbiol. Ecol. 2019, 95, 203. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Bacigalupe, R.; García-Carral, C.; Boix-Amoros, A.; Argüello, H.; Silva, C.B.; de Los Angeles Checa, M.; Mira, A.; Rodriguez, J.M. Microbiota of human precolostrum and its potential role as a source of bacteria to the infant mouth. Sci. Rep. 2019, 9, 8435. [Google Scholar] [CrossRef] [PubMed]
- Meale, S.J.; Li, S.; Azevedo, P.; Derakhshani, H.; Plaizier, J.C.; Khafipour, E.; Steele, M.A. Development of ruminal and fecal microbiomes are affected by weaning but not weaning strategy in dairy calves. Front. Microbiol. 2016, 7, 582. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Gomez, A.; White, B.; Loften, J.; Drackley, J. Changes in the intestinal bacterial community, short-chain fatty acid profile, and intestinal development of preweaned Holstein calves. 2. Effects of gastrointestinal site and age. J. Dairy Sci. 2016, 99, 9703–9715. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynönen, U.; Iivanainen, A.; Niku, M. The compo-sition of the perinatal intestinal microbiota in cattle. Sci. Rep. 2018, 8, 1–14. [Google Scholar]
- Dill-McFarland, K.A.; Weimer, P.J.; Breaker, J.D.; Suen, G. Diet Influences Early Microbiota Development in Dairy Calves without Long-Term Impacts on Milk Production. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, G.; Teixeira, A.G.V.; Foditsch, C.; Bicalho, M.L.; Machado, V.S.; Bicalho, R.C. Fecal Microbial Diversity in Pre-Weaned Dairy Calves as Described by Pyrosequencing of Metagenomic 16S rDNA. Associations of Faecalibacterium Species with Health and Growth. PLoS ONE 2013, 8, e63157. [Google Scholar] [CrossRef] [PubMed]
- Dill-McFarland, K.A.; Breaker, J.D.; Suen, G. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 2017, 7, 40864. [Google Scholar] [CrossRef]
- Klein-Jöbstl, D.; Schornsteiner, E.; Mann, E.; Wagner, M.; Drillich, M.; Schmitz-Esser, S. Pyrosequencing reveals diverse fecal microbiota in Simmental calves during early development. Front. Microbiol. 2014, 5, 622. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Arroyo, L.; Costa, M.; Viel, L.; Weese, J. Characterization of the Fecal Bacterial Microbiota of Healthy and Diarrheic Dairy Calves. J. Vet. Intern. Med. 2017, 31, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhu, H.; Qiu, P. Aging progression of human gut microbiota. BMC Microbiol. 2019, 19, 236. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.; Marcondes, M.I.; Noronha, M.F.; Resende, R.T.; Machado, F.S.; Mantovani, H.C.; Dill-McFarland, K.A.; Suen, G. Effect of Pre-weaning Diet on the Ruminal Archaeal, Bacterial, and Fungal Communities of Dairy Calves. Front. Microbiol. 2017, 8, 1553. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, E.; Kelly, A.; McCabe, M.S.; Kenny, D.A.; Waters, S.M. Effect of a butyrate-fortified milk replacer on gastrointestinal microbiota and products of fermentation in artificially reared dairy calves at weaning. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Ma, T.; Villot, C.; Renaud, D.; Skidmore, A.; Chevaux, E.; Steele, M. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: Prediction of diarrhea. ISME J. 2020, 14, 2223–2235. [Google Scholar] [CrossRef]
- Kim, H.S.; Whon, T.W.; Sung, H.; Jeong, Y.-S.; Jung, E.S.; Shin, N.-R.; Hyun, D.-W.; Kim, P.S.; Lee, J.-Y.; Lee, C.H. Longi-tudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 2021, 12, 1–16. [Google Scholar]
- Bartels, C.J.; Holzhauer, M.; Jorritsma, R.; Swart, W.A.; Lam, T.J. Prevalence, prediction and risk factors of enteropathogens in normal and non-normal faeces of young Dutch dairy calves. Prev. Vet. Med. 2010, 93, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zeng, D.; Wang, Q.; Wang, N.; Zeng, B.; Niu, L.; Ni, X. Diarrhea-associated intestinal microbiota in captive Sichuan golden snub-nosed monkeys (Rhinopithecus roxellana). Microbes Environ. 2018, 33, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Suchodolski, J.S.; Foster, M.L.; Sohail, M.U.; Leutenegger, C.; Queen, E.V.; Steiner, J.M.; Marks, S.L. The Fecal Microbiome in Cats with Diarrhea. PLoS ONE 2015, 10, e0127378. [Google Scholar] [CrossRef]
- Guard, B.C.; Barr, J.W.; Reddivari, L.; Klemashevich, C.; Jayaraman, A.; Steiner, J.M.; Vanamala, J.; Suchodolski, J.S. Characterization of microbial dysbiosis and metabolomic changes in dogs with acute diarrhea. PLoS ONE 2015, 10, e0127259. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Bry, L.; Falk, P.G.; Midtvedt, T.; Gordon, J.I. A model of host-microbial interactions in an open mammalian ecosystem. Science 1996, 273, 1380–1383. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Donaldson, G.P.; Mikulski, Z.; Boyajian, S.; Ley, K.; Mazmanian, S.K. Bacterial colonization factors control specificity and stability of the gut microbiota. Nat. Cell Biol. 2013, 501, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.; Smith, G.W. Pathophysiology of Diarrhea in Calves. Vet. Clin. N. Am. Food Anim. Pr. 2009, 25, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Su, J.; Zheng, X.; Wang, C.; Chen, S.; Liu, J.; Lv, Y.; Fan, S.; Zhao, A.; et al. Age-related compositional changes and correlations of gut microbiome, serum metabolome, and immune factor in rats. Geroscience 2020, 1–17. [Google Scholar] [CrossRef]
- Ichikawa-Seki, M.; Motooka, D.; Kinami, A.; Murakoshi, F.; Takahashi, Y.; Aita, J.; Hayashi, K.; Tashibu, A.; Nakamura, S.; Iida, T.; et al. Specific increase of Fusobacterium in the faecal microbiota of neonatal calves infected with Cryptosporidium parvum. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Fecteau, M.-E.; Pitta, D.W.; Vecchiarelli, B.; Indugu, N.; Kumar, S.; Gallagher, S.C.; Fyock, T.L.; Sweeney, R.W. Dysbiosis of the Fecal Microbiota in Cattle Infected with Mycobacterium avium subsp. paratuberculosis. PLoS ONE 2016, 11, e0160353. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, M.; Aldridge, B.; Lowe, J. Dysbiosis of the fecal microbiota in feedlot cattle with hemorrhagic diarrhea. Microb. Pathog. 2018, 115, 123–130. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name 1 | Total Bases 2 | Read Count 3 | Q20(%) 4 | Sample Name 1 | Total Bases 2 | Read Count 3 | Q20(%) 4 |
|---|---|---|---|---|---|---|---|
| 18014.0 wk 5 | 68,907,327 | 151,588 | 97.31 | 1802.5 wk 5 | 120,851,766 | 266,565 | 98.53 |
| 18014.1 wk | 73,276,256 | 161,129 | 97.88 | 1802.6 wk | 121,996,008 | 271,757 | 98.59 |
| 18014.2 wk | 75,203,142 | 164,647 | 97.38 | 1802.7 wk | 127,730,965 | 283,648 | 98.43 |
| 18014.3 wk | 50,686,786 | 111,548 | 97.43 | 1802.8 wk | 110,898,306 | 246,337 | 98.49 |
| 18014.4 wk | 65,018,120 | 143,310 | 97.36 | 1803.0 wk | 27,571,311 | 60,529 | 97.35 |
| 18014.5 wk | 58,783,722 | 129,694 | 97.51 | 1803.1 wk | 58,760,407 | 128,844 | 97.45 |
| 18014.6 wk | 55,635,573 | 123,136 | 97.55 | 1803.2 wk | 75,370,647 | 166,826 | 97.62 |
| 18014.7 wk | 46,376,877 | 102,750 | 97.67 | 1803.3 wk | 62,276,136 | 137,975 | 97.6 |
| 18014.8 wk | 51,792,102 | 115,213 | 97.5 | 1803.4 wk | 77,599,897 | 172,809 | 97.55 |
| 18030.0 wk | 86,128,455 | 191,030 | 98.48 | 1803.5 wk | 52,503,714 | 116,060 | 97.49 |
| 18030.1 wk | 73,886,165 | 163,363 | 98.35 | 1803.6 wk | 56,767,208 | 125,325 | 97.48 |
| 18030.2 wk | 60,748,672 | 134,589 | 98.53 | 1803.7 wk | 56,836,785 | 126,461 | 97.6 |
| 18030.3 wk | 76,840,598 | 169,738 | 98.32 | 1803.8 wk | 62,833,274 | 139,711 | 97.65 |
| 18030.4 wk | 81,039,398 | 178,623 | 98.36 | 1809.0 wk | 66,532,385 | 146,064 | 98.92 |
| 18030.5 wk | 64,971,503 | 143,031 | 97.74 | 1809.1 wk | 71,049,665 | 154,645 | 98.66 |
| 18030.6 wk | 62,737,787 | 139,009 | 98.22 | 1809.2 wk | 79,287,856 | 173,699 | 98.69 |
| 18030.7 wk | 88,812,047 | 197,187 | 98.38 | 1809.3 wk | 69,925,292 | 152,643 | 98.87 |
| 18030.8 wk | 66,357,601 | 147,718 | 98.39 | 1809.4 wk | 71,951,762 | 158,274 | 98.68 |
| 1802.0 wk | 101,856,946 | 223,217 | 98.29 | 1809.5 wk | 73,684,048 | 161,208 | 98.81 |
| 1802.1 wk | 126,700,114 | 280,737 | 98.66 | 1809.6 wk | 70,792,739 | 155,381 | 98.5 |
| 1802.2 wk | 126,537,869 | 276,213 | 98.59 | 1809.7 wk | 71,128,878 | 156,248 | 98.51 |
| 1802.3 wk | 120,722,588 | 265,181 | 98.61 | 1809.8 wk | 73,935,709 | 164,995 | 98.46 |
| 1802.4 wk | 144,618,157 | 319,434 | 98.81 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.-T.; Lee, S.-J.; Kim, T.-Y.; Lee, H.-G.; Atikur, R.M.; Gu, B.-H.; Kim, D.-H.; Park, B.-Y.; Son, J.-K.; Kim, M.-H. Dynamic Changes in Fecal Microbial Communities of Neonatal Dairy Calves by Aging and Diarrhea. Animals 2021, 11, 1113. https://doi.org/10.3390/ani11041113
Kim E-T, Lee S-J, Kim T-Y, Lee H-G, Atikur RM, Gu B-H, Kim D-H, Park B-Y, Son J-K, Kim M-H. Dynamic Changes in Fecal Microbial Communities of Neonatal Dairy Calves by Aging and Diarrhea. Animals. 2021; 11(4):1113. https://doi.org/10.3390/ani11041113
Chicago/Turabian StyleKim, Eun-Tae, Sang-Jin Lee, Tae-Yong Kim, Hyo-Gun Lee, Rahman M. Atikur, Bon-Hee Gu, Dong-Hyeon Kim, Beom-Young Park, Jun-Kyu Son, and Myung-Hoo Kim. 2021. "Dynamic Changes in Fecal Microbial Communities of Neonatal Dairy Calves by Aging and Diarrhea" Animals 11, no. 4: 1113. https://doi.org/10.3390/ani11041113
APA StyleKim, E.-T., Lee, S.-J., Kim, T.-Y., Lee, H.-G., Atikur, R. M., Gu, B.-H., Kim, D.-H., Park, B.-Y., Son, J.-K., & Kim, M.-H. (2021). Dynamic Changes in Fecal Microbial Communities of Neonatal Dairy Calves by Aging and Diarrhea. Animals, 11(4), 1113. https://doi.org/10.3390/ani11041113