
Genotype Imputation to Improve the Cost-Efficiency of Genomic Selection in Rabbits

 ,
,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Simulation Structure
2.2. Imputation Strategies
2.3. Estimating Breeding Values and Response to Genomic Selection
3. Results
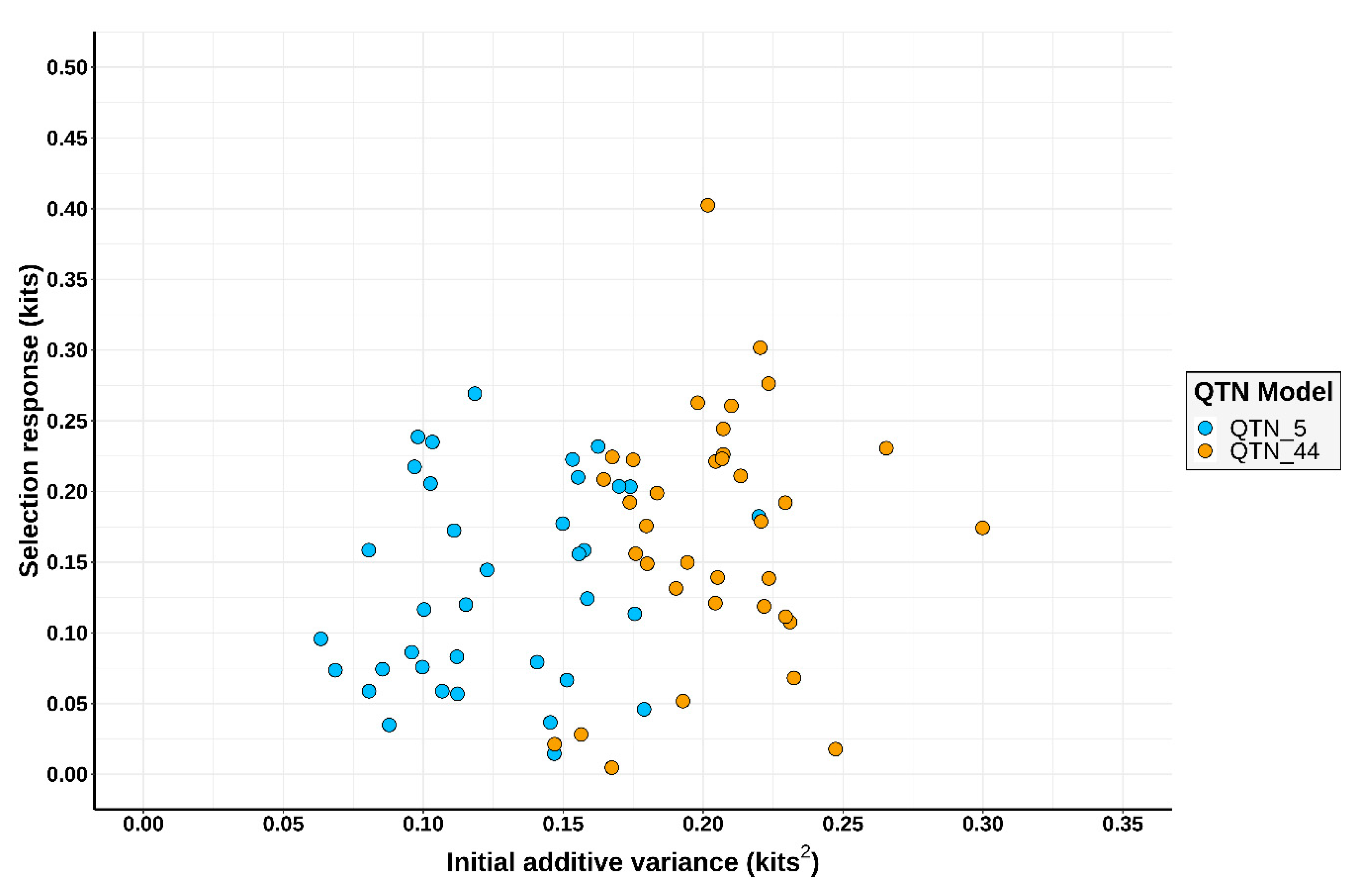
3.1. Simulation Outcomes
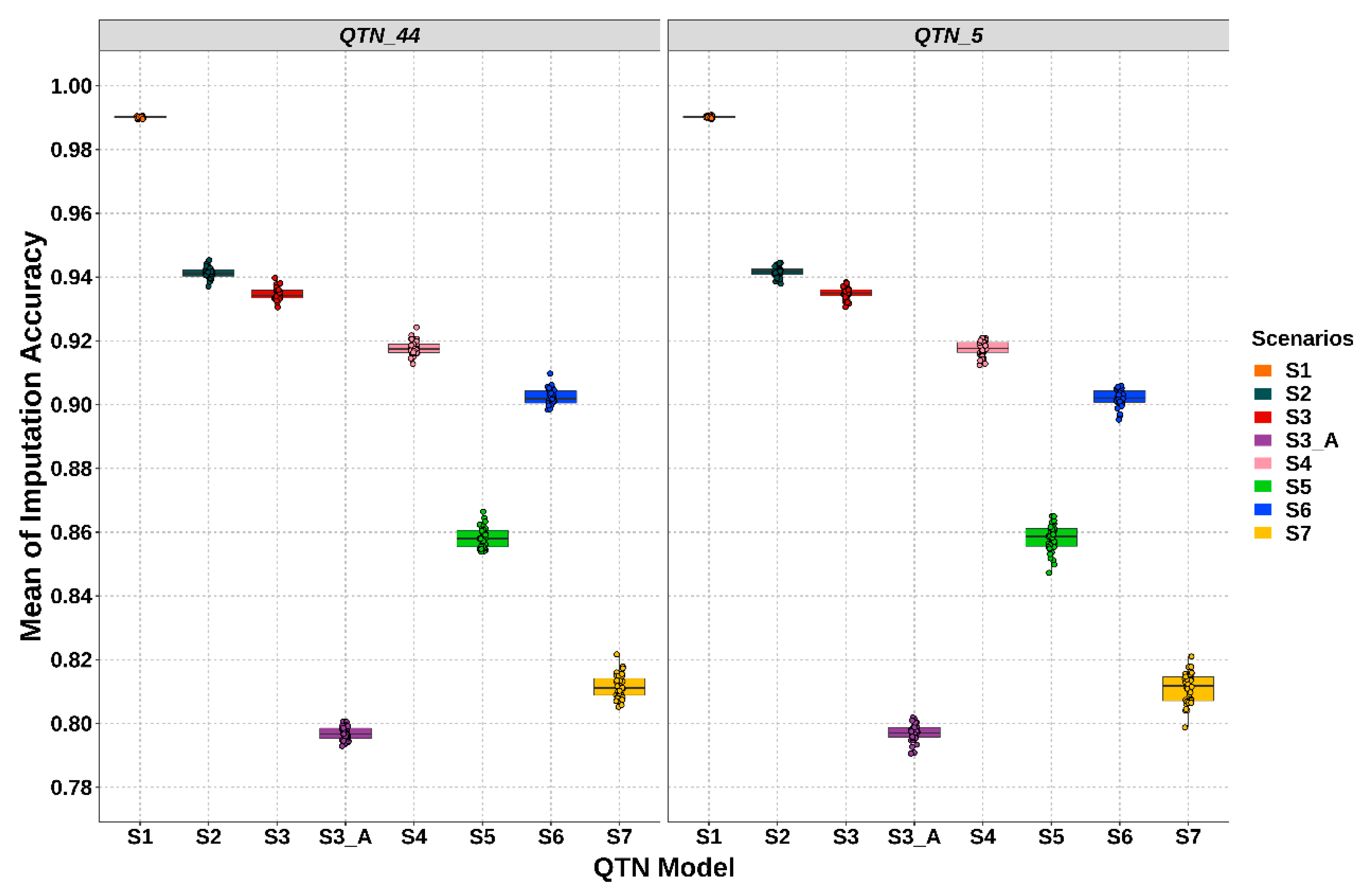
3.2. Performance of Imputation Strategies
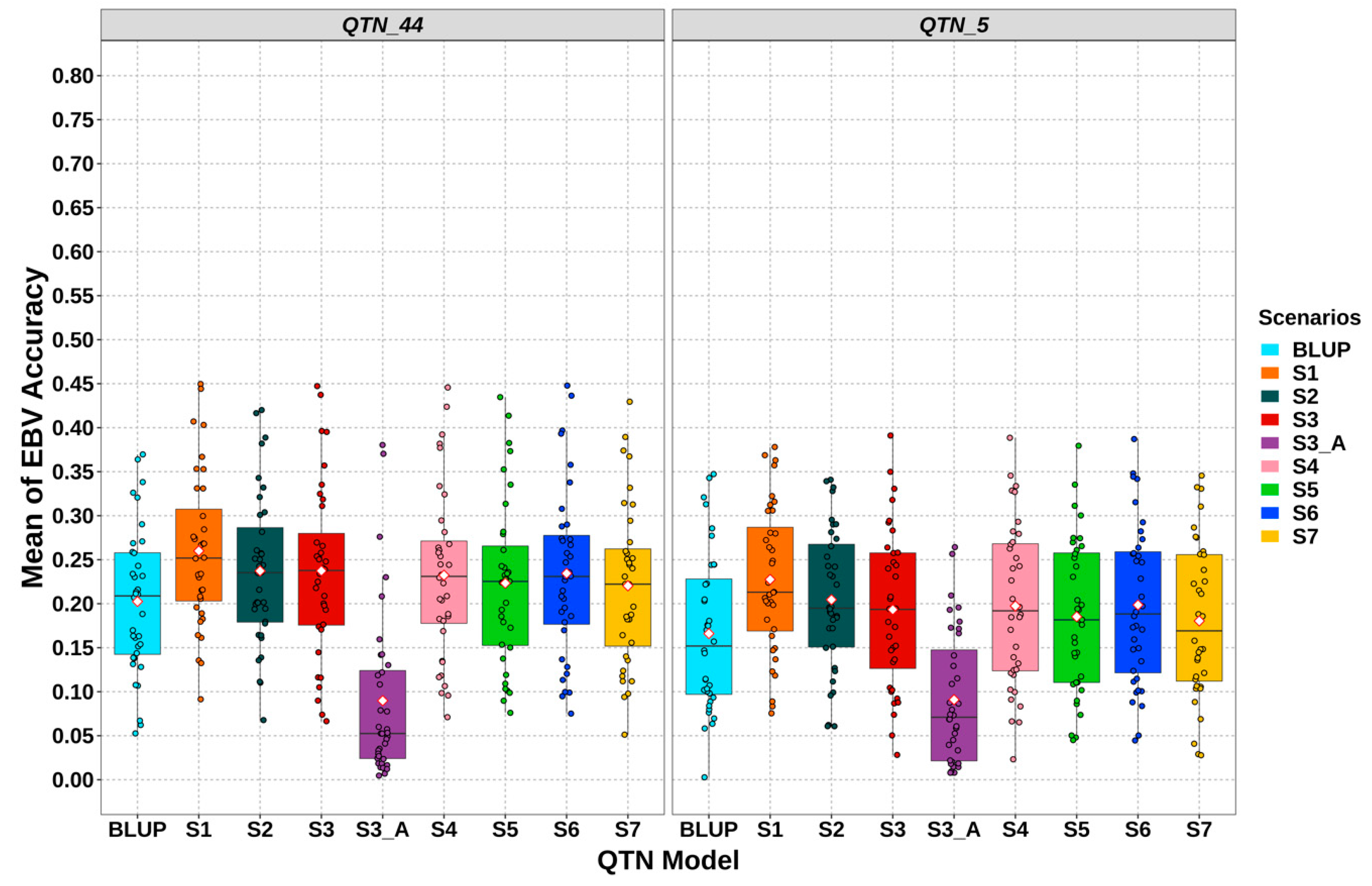
3.3. Gemomic Prediction vs. Pedigree-Based Analyses
3.4. Genotyping Costs
4. Discussion
4.1. Imputation Strategies
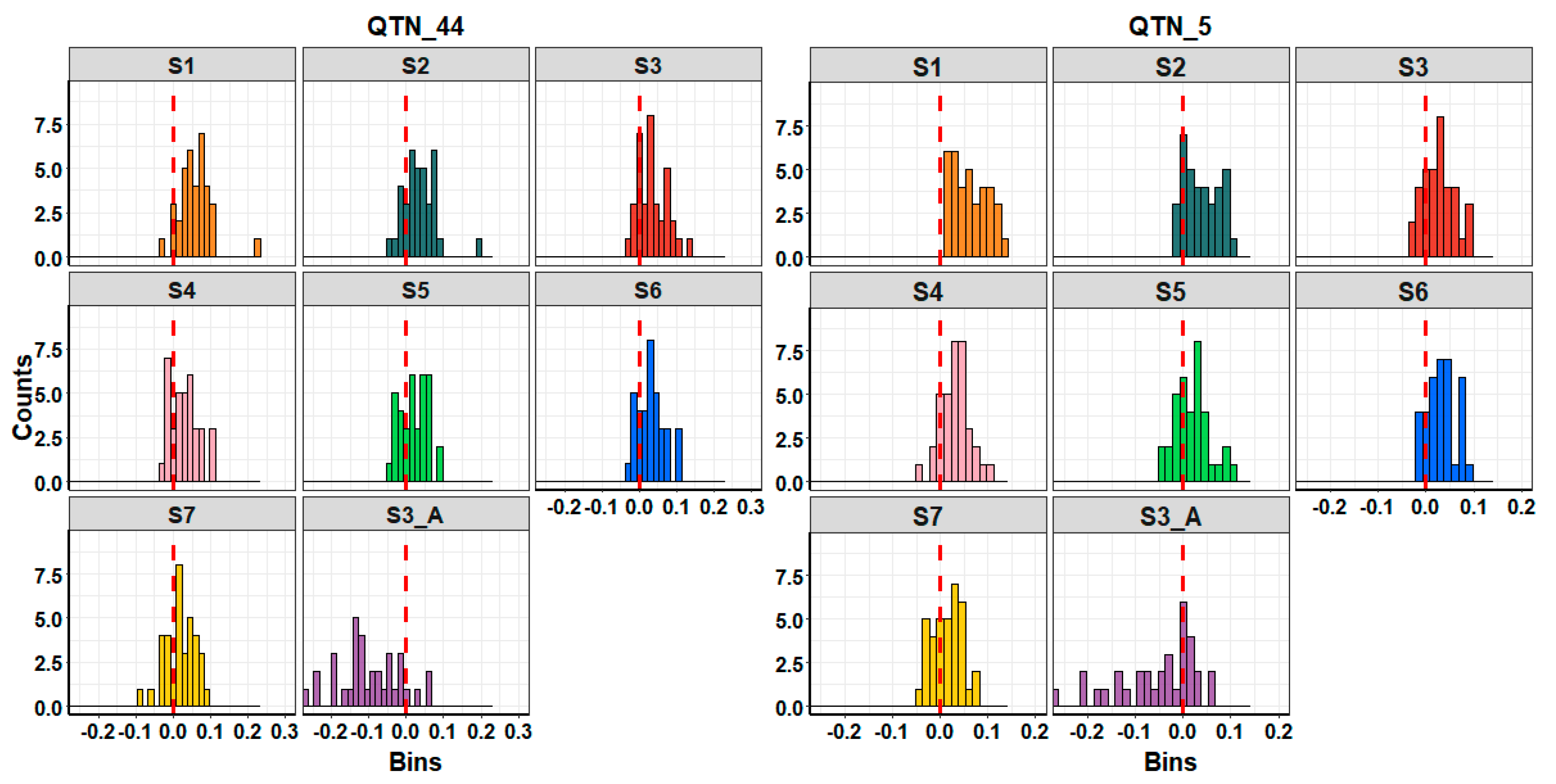
4.2. Causes That Affect Genomic Prediction
4.3. Comparison with Other Studies
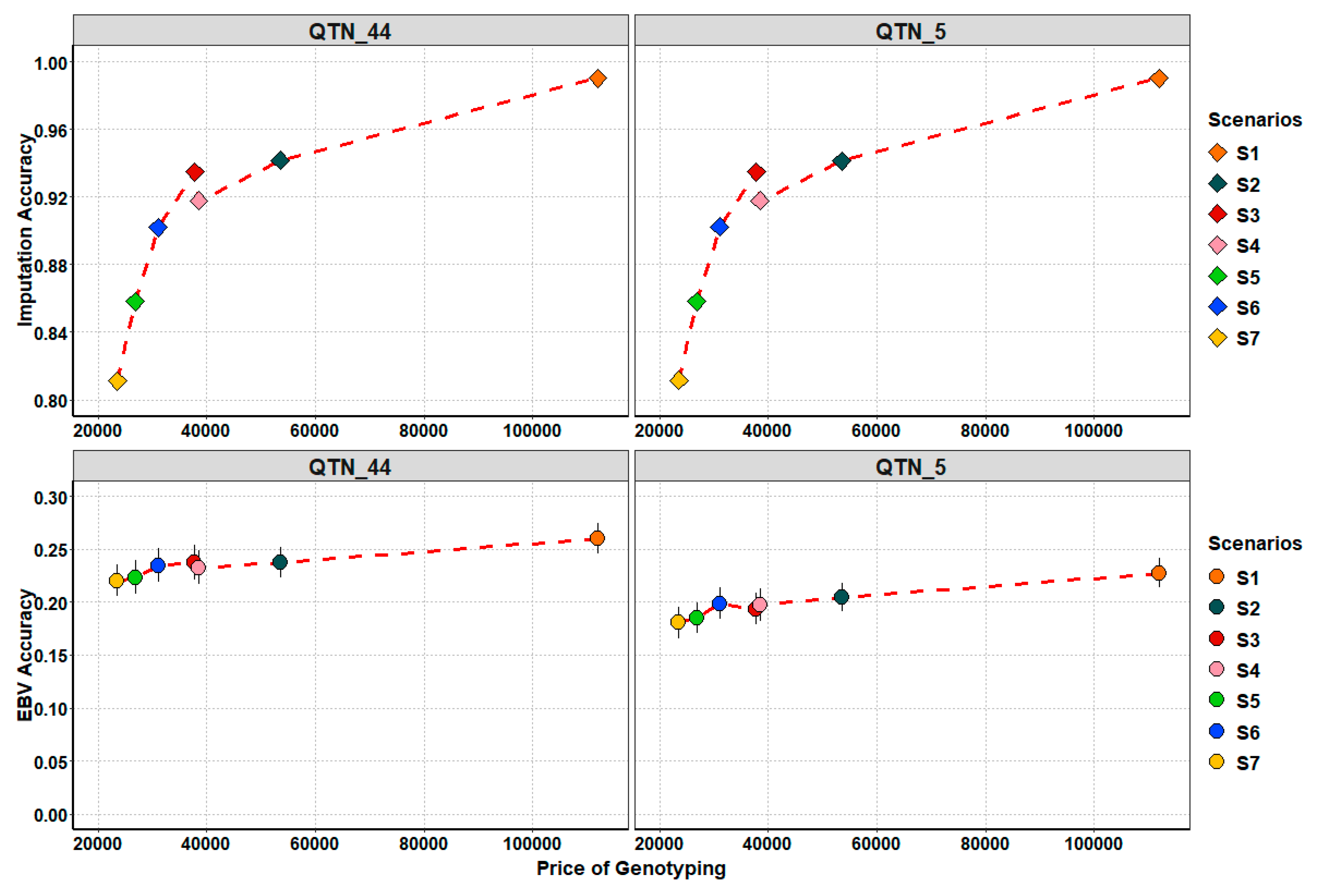
4.4. Searching for Trade-Off: Cost and Genetic Accuracy Trends
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cullere, M.; Dalle Zotte, A. Rabbit meat production and consumption: State of knowledge and future perspectives. Meat Sci. 2018, 143, 137–146. [Google Scholar] [CrossRef]
- European Commission. Overview Report: Commercial Farming of Rabbits in the European Union. Luxemb. Publ. Off. Eur. Union 2017, 16. [Google Scholar] [CrossRef]
- Cartuche, L.; Pascual, M.; Gómez, E.A.; Blasco, A. Economic weights in rabbit meat production. World Rabbit Sci. 2014, 22, 165–177. [Google Scholar] [CrossRef]
- Baselga, M. Genetic Improvement of Meat Rabbits. Programmes and Diffusion. In Proceedings of the 8th World Rabbit Congress, Puebla, Mexico, 7–14 September 2004; pp. 1–13. [Google Scholar]
- Fernández, E.N.; Sánchez, J.P.; Martínez, R.; Legarra, A.; Baselga, M. Role of inbreeding depression, non-inbred dominance deviations and random year-season effect in genetic trends for prolificacy in closed rabbit lines. J. Anim. Breed. Genet. 2017, 134, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Ragab, M.; Baselga, M. A comparison of reproductive traits of four maternal lines of rabbits selected for litter size at weaning and founded on different criteria. Livest. Sci. 2011, 136, 201–206. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6. [Google Scholar] [CrossRef]
- da Cruz, V.A.R.; Brito, L.F.; Schenkel, F.S.; de Oliveira, H.R.; Jafarikia, M.; Feng, Z. Strategies for within-litter selection of piglets using ultra-low density SNP panels. Livest. Sci. 2019, 220, 173–179. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Dekkers, J.C.M. The impact of genetic relationship information on genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef]
- Jattawa, D.; Elzo, M.A.; Koonawootrittriron, S.; Suwanasopee, T. Imputation Accuracy from Low to Moderate Density Single Nucleotide Polymorphism Chips in a Thai Multibreed Dairy Cattle Population. Asian-Australas. J. Anim. Sci. 2016, 29, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Schöpke, K.; Swalve, H.H. Review: Opportunities and challenges for small populations of dairy cattle in the era of genomics. Animal 2016, 10, 1050–1060. [Google Scholar] [CrossRef]
- Wolc, A.; Kranis, A.; Arango, J.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Avendano, A.; Watson, K.A.; Hickey, J.M.; de los Campos, G.; et al. Implementation of genomic selection in the poultry industry. Anim. Front. 2016, 6, 23–31. [Google Scholar] [CrossRef]
- Knol, E.F.; Nielsen, B.; Knap, P.W. Genomic selection in commercial pig breeding. Anim. Front. 2016, 6, 15–22. [Google Scholar] [CrossRef]
- Garrick, D.J. The role of genomics in pig improvement. Anim. Prod. Sci. 2017, 57, 2360–2365. [Google Scholar] [CrossRef]
- Ibáñez-Escriche, N.; Forni, S.; Noguera, J.L.; Varona, L. Genomic information in pig breeding: Science meets industry needs. Livest. Sci. 2014, 166, 94–100. [Google Scholar] [CrossRef]
- Blasco, A.; Pena, R.N. Current Status of Genomic Maps: Genomic Selection/GBV in Livestock. In Animal Biotechnology 2: Emerging Breeding Technologies; Niemann, H., Wrenzycki, C., Eds.; Springer International Publishing: Cham, Germany, 2018; pp. 61–80. ISBN 978-3-319-92348-2. [Google Scholar]
- Ibáñez-Escriche, N.; González-Recio, O. Review. Promises, pitfalls and challenges of genomic selection in breeding programs. Span. J. Agric. Res. 2011, 9, 404–413. [Google Scholar] [CrossRef]
- Cleveland, M.A.; Hickey, J.M. Practical implementation of cost-effective genomic selection in commercial pig breeding using imputation1. J. Anim. Sci. 2013, 91, 3583–3592. [Google Scholar] [CrossRef]
- Grossi, D.A.; Brito, L.F.; Jafarikia, M.; Schenkel, F.S.; Feng, Z. Genotype imputation from various low-density SNP panels and its impact on accuracy of genomic breeding values in pigs. Animal 2018, 12, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.C.; Judge, M.M.; Fair, S.; Berry, D.P. High imputation accuracy from informative low-to-medium density single nucleotide polymorphism genotypes is achievable in sheep. J. Anim. Sci. 2019, 97, 1550–1567. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, G.; Li, C.; Stothard, P. Genotype Imputation Methods and Their Effects on Genomic Predictions in Cattle. Springer Sci. Rev. 2016, 4, 79–98. [Google Scholar] [CrossRef]
- Calus, M.P.L.; Bouwman, A.C.; Hickey, J.M.; Veerkamp, R.F.; Mulder, H.A. Evaluation of measures of correctness of genotype imputation in the context of genomic prediction: A review of livestock applications. Animal 2014, 8, 1743–1753. [Google Scholar] [CrossRef]
- Faux, A.-M.; Gorjanc, G.; Gaynor, R.C.; Battagin, M.; Edwards, S.M.; Wilson, D.L.; Hearne, S.J.; Gonen, S.; Hickey, J.M. AlphaSim: Software for Breeding Program Simulation. Plant. Genome 2016, 9. [Google Scholar] [CrossRef]
- Chen, G.K.; Marjoram, P.; Wall, J.D. Fast and flexible Simulation of DNA Sequence Data. Genome Res. 2009, 19, 136–142. [Google Scholar] [CrossRef]
- García, M.L.; Baselga, M. Estimation of Genetic Response to selection in litter size of rabbits using a cryopreserved control population. Livest. Prod. Sci. 2002, 74, 45–53. [Google Scholar] [CrossRef]
- Ragab, M.; Sánchez, J.P.; Mínguez, C.; Vicente, J.S.; Baselga, M. Litter size components in a full diallel cross of four maternal lines of rabbits. J. Anim. Sci. 2014, 92, 3231–3236. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Argente, M.J. Major Components in Limiting Litter Size. In Insights from Animal Reproduction; Payan Carreira, R., Ed.; InTech: London, UK, 2016; pp. 87–114. ISBN 978-953-51-2268-5. [Google Scholar]
- R Core Team. R: A Language and Environmental for Statistical Computing; R foundation for statistical computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 15 August 2020).
- Hickey, J.M.; Kinghorn, B.P.; Tier, B.; van der Werf, J.H.; Cleveland, M.A. A phasing and imputation method for pedigreed populations that results in a single-stage genomic evaluation. Genet. Sel. Evol. 2012, 44, 9. [Google Scholar] [CrossRef]
- Hickey, J.M.; Crossa, J.; Babu, R.; de los Campos, G. Factors Affecting the Accuracy of Genotype Imputation in Populations from Several Maize Breeding Programs. Crop. Sci. 2012, 52, 654–663. [Google Scholar] [CrossRef]
- Legarra, A.; Christensen, O.F.; Aguilar, I.; Misztal, I. Single Step, a general approach for genomic selection. Livest. Sci. 2014, 166, 54–65. [Google Scholar] [CrossRef]
- Legarra, A.; Aguilar, I.; Misztal, I. A relationship matrix including full pedigree and genomic information. J. Dairy Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef]
- Christensen, O.F.; Lund, M.S. Genomic prediction when some animals are not genotyped. Genet. Sel. Evol. 2010, 42, 2. [Google Scholar] [CrossRef]
- Calus, M.P.L. Editorial: Genomic selection with numerically small reference populations. Animal 2016, 10, 1016–1017. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Garcia-Baccino, C.A.; Legarra, A.; Christensen, O.F.; Misztal, I.; Pocrnic, I.; Vitezica, Z.G.; Cantet, R.J.C. Metafounders are related to Fst fixation indices and reduce bias in single-step genomic evaluations. Genet. Sel. Evol. 2017, 49, 34. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, I.; Fernandez, E.N.; Blasco, A.; Ravagnolo, O.; Legarra, A. Effects of ignoring inbreeding in model-based accuracy for BLUP and SSGBLUP. J. Anim. Breed. Genet. 2020, 137, 356–364. [Google Scholar] [CrossRef]
- Aguilar, I.; Tsuruta, S.; Masuda, Y.; Lourenco, D.A.L.; Legarra, A.; Misztal, I. BLUPF90 suite of programs for animal breeding. In Proceedings of the 11th World Congress of Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018; p. 11.751. [Google Scholar]
- Pszczola, M.; Strabel, T.; van Arendonk, J.A.M.; Calus, M.P.L. The impact of genotyping different groups of animals on accuracy when moving from traditional to genomic selection. J. Dairy Sci. 2012, 95, 5412–5421. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, J.; Tsuruta, S.; Lourenco, D.; Masuda, Y.; Huang, Y.; Gray, K.A.; Misztal, I. Changes in genetic parameters for fitness and growth traits in pigs under genomic selection. J. Anim. Sci. 2020, 98, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Willer, C.; Sanna, S.; Abecasis, G. Genotype imputation. Annu. Rev. Genom. Hum. Genet. 2009, 10, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Gorjanc, G.; Battagin, M.; Dumasy, J.; Antolin, R.; Gaynor, R.C.; Hickey, J.M. Prospects for Cost-Effective Genomic Selection via Accurate Within-Family Imputation. Crop Sci. 2017, 57, 216–228. [Google Scholar] [CrossRef]
- Tsai, H.Y.; Matika, O.; Edwards, S.M.K.; Antolín-Sánchez, R.; Hamilton, A.; Guy, D.R.; Tinch, A.E.; Gharbi, K.; Stear, M.J.; Taggart, J.B.; et al. Genotype imputation to improve the cost-efficiency of genomic selection in farmed Atlantic salmon. G3 Genes Genomes Genet. 2017, 7, 1377–1383. [Google Scholar] [CrossRef]
- Lopez, B.I.; Lee, S.H.; Shin, D.H.; Oh, J.D.; Chai, H.H.; Park, W.; Park, J.E.; Lim, D. Accuracy of genomic evaluation using imputed high-density genotypes for carcass traits in commercial Hanwoo population. Livest. Sci. 2020, 241, 104256. [Google Scholar] [CrossRef]
- Tabor, H.K.; Risch, N.J.; Myers, R.M. Candidate-gene approaches for studying complex genetic traits: Practical considerations. Nat. Rev. Genet. 2002, 3, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Weigel, K.A.; de los Campos, G.; Vazquez, A.I.; Rosa, G.J.M.; Gianola, D.; Van Tassell, C.P. Accuracy of direct genomic values derived from imputed single nucleotide polymorphism genotypes in Jersey cattle. J. Dairy Sci. 2010, 93, 5423–5435. [Google Scholar] [CrossRef]
- Pimentel, E.C.G.; Wensch-Dorendorf, M.; König, S.; Swalve, H.H. Enlarging a training set for genomic selection by imputation of un-genotyped animals in populations of varying genetic architecture. Genet. Sel. Evol. 2013, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.A.; Silva, F.F.; Silva, D.A.; Silva, H.T.; Costa, C.N.; Lopes, P.S.; Veroneze, R.; Thompson, G.; Carvalheira, J. Genotype imputation strategies for Portuguese Holstein cattle using different SNP panels. Czech. J. Anim. Sci. 2019, 64, 377–386. [Google Scholar] [CrossRef]
- Zhang, Z.; Druet, T. Marker imputation with low-density marker panels in Dutch Holstein cattle. J. Dairy Sci. 2010, 93, 5487–5494. [Google Scholar] [CrossRef] [PubMed]
- Heidaritabar, M.; Calus, M.P.L.; Vereijken, A.; Groenen, M.A.M.; Bastiaansen, J.W.M. Accuracy of imputation using the most common sires as reference population in layer chickens. BMC Genet. 2015, 16, 1–14. [Google Scholar] [CrossRef]
- Bouwman, A.C.; Hickey, J.M.; Calus, M.P.; Veerkamp, R.F. Imputation of non-genotyped individuals based on genotyped relatives: Assessing the imputation accuracy of a real case scenario in dairy cattle. Genet. Sel. Evol. 2014, 46, 1–11. [Google Scholar] [CrossRef]
- Misztal, I.; Aggrey, S.E.; Muir, W.M. Experiences with a single-step genome evaluation. Poult. Sci. 2013, 92, 2530–2534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yin, L.; Wang, M.; Yuan, X.; Liu, X. Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Garrick, D.J. Genomic BLUP Decoded: A Look into the Black Box of Genomic Prediction. Genetics 2013, 194, 597–607. [Google Scholar] [CrossRef]
- Fragomeni, B.O.; Lourenco, D.A.L.; Masuda, Y.; Legarra, A.; Misztal, I. Incorporation of causative quantitative trait nucleotides in single-step GBLUP. Genet. Sel. Evol. 2017, 49, 59. [Google Scholar] [CrossRef]
- Zhang, X.; Lourenco, D.; Aguilar, I.; Legarra, A.; Misztal, I. Weighting Strategies for Single-Step Genomic BLUP: An Iterative Approach for Accurate Calculation of GEBV and GWAS. Front. Genet. 2016, 7, 151. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.L.; Dekkers, J.C.M.; Garrick, D.J. A class of Bayesian methods to combine large numbers of genotyped and non-genotyped animals for whole-genome analyses. Genet. Sel. Evol. 2014, 46, 50. [Google Scholar] [CrossRef]
- Song, H.; Ye, S.; Jiang, Y.; Zhang, Z.; Zhang, Q.; Ding, X. Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs. Genet. Sel. Evol. 2019, 51, 58. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, E.C.G.; Edel, C.; Emmerling, R.; Götz, K.-U. How imputation errors bias genomic predictions. J. Dairy Sci. 2015, 98, 4131–4138. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Imputation Strategy | Training Populations | For Imputation | Validated Population (Genomic Prediction) | |||
|---|---|---|---|---|---|---|
| 26th Generation | 27th Generation | 28th Generation | ||||
| Grand-Dams (150) | Grand-Sires (35) | Dams (150) | Sires (35) | Progeny (1500) | Progeny (1500) | |
| S1 | HD | HD | HD | HD | MD | i-HD |
| S2 | HD | HD | HD | HD | LD | i-HD |
| S3 | MD | HD | HD | HD | ½ LD | ½ i-HD + ½ NG |
| S4 | MD | HD | MD | HD | LD | i-HD |
| S5 | LD | HD | LD | HD | LD | i-HD |
| S6 | NG | HD | MD | HD | LD | i-HD |
| S7 | NG | HD | NG | HD | LD | i-HD |
| S3_A | MD | HD | HD | HD | ½ LD | ½ i-HD + ½ i-WG |
| Generation | Pedigree | Phenotypic 1 | Genomic 2 |
|---|---|---|---|
| 23th | 300 | 150 | 0:0 |
| 24th | 300 | 150 | 0:0 |
| 25th | 300 | 150 | 0:0 |
| 26th | 300 | 150 | 35:150 |
| 27th | 300 | 150 | 35:150 |
| 28th | 1500 | 0 | 0:1500 |
| Scenario | Selection Response 1 | SE-1 2 | Percentage of ACS 3 | SE-2 4 |
|---|---|---|---|---|
| BLUP | 0.105 | 0.007 | 27.81 | 0.56 |
| S1 | 0.129 | 0.007 | 30.54 | 0.56 |
| S2 | 0.120 | 0.007 | 29.45 | 0.54 |
| S3 | 0.117 | 0.008 | 29.36 | 0.60 |
| S3_A | 0.046 | 0.008 | 23.62 | 0.65 |
| S4 | 0.114 | 0.007 | 28.93 | 0.54 |
| S5 | 0.108 | 0.007 | 28.42 | 0.53 |
| S6 | 0.116 | 0.007 | 29.06 | 0.55 |
| S7 | 0.109 | 0.007 | 28.26 | 0.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancin, E.; Sosa-Madrid, B.S.; Blasco, A.; Ibáñez-Escriche, N. Genotype Imputation to Improve the Cost-Efficiency of Genomic Selection in Rabbits. Animals 2021, 11, 803. https://doi.org/10.3390/ani11030803
Mancin E, Sosa-Madrid BS, Blasco A, Ibáñez-Escriche N. Genotype Imputation to Improve the Cost-Efficiency of Genomic Selection in Rabbits. Animals. 2021; 11(3):803. https://doi.org/10.3390/ani11030803
Chicago/Turabian StyleMancin, Enrico, Bolívar Samuel Sosa-Madrid, Agustín Blasco, and Noelia Ibáñez-Escriche. 2021. "Genotype Imputation to Improve the Cost-Efficiency of Genomic Selection in Rabbits" Animals 11, no. 3: 803. https://doi.org/10.3390/ani11030803
APA StyleMancin, E., Sosa-Madrid, B. S., Blasco, A., & Ibáñez-Escriche, N. (2021). Genotype Imputation to Improve the Cost-Efficiency of Genomic Selection in Rabbits. Animals, 11(3), 803. https://doi.org/10.3390/ani11030803