
A Comparison of the Colonic Microbiome and Volatile Organic Compound Metabolome of Anoplocephala perfoliata Infected and Non-Infected Horses: A Pilot Study


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Microbiome Profiling: 16S rRNA Sequencing
2.3. Volatile Organic Compound (VOC) Metabolome Profiling
2.4. Data Analysis
2.4.1. Microbiome
2.4.2. Volatile Organic Compound (VOC) Metabolome
2.5. Integration of Omics Data
3. Results
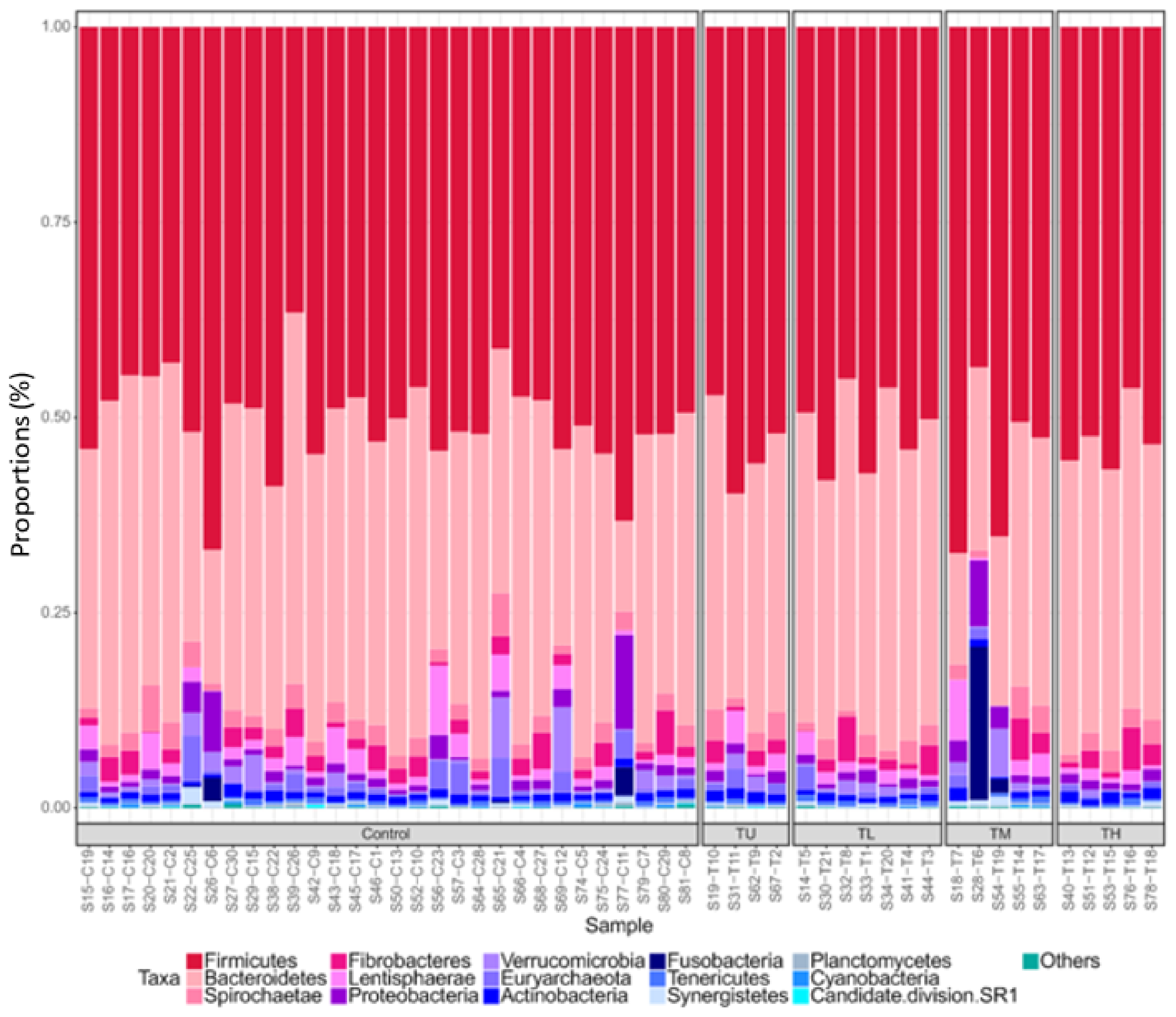
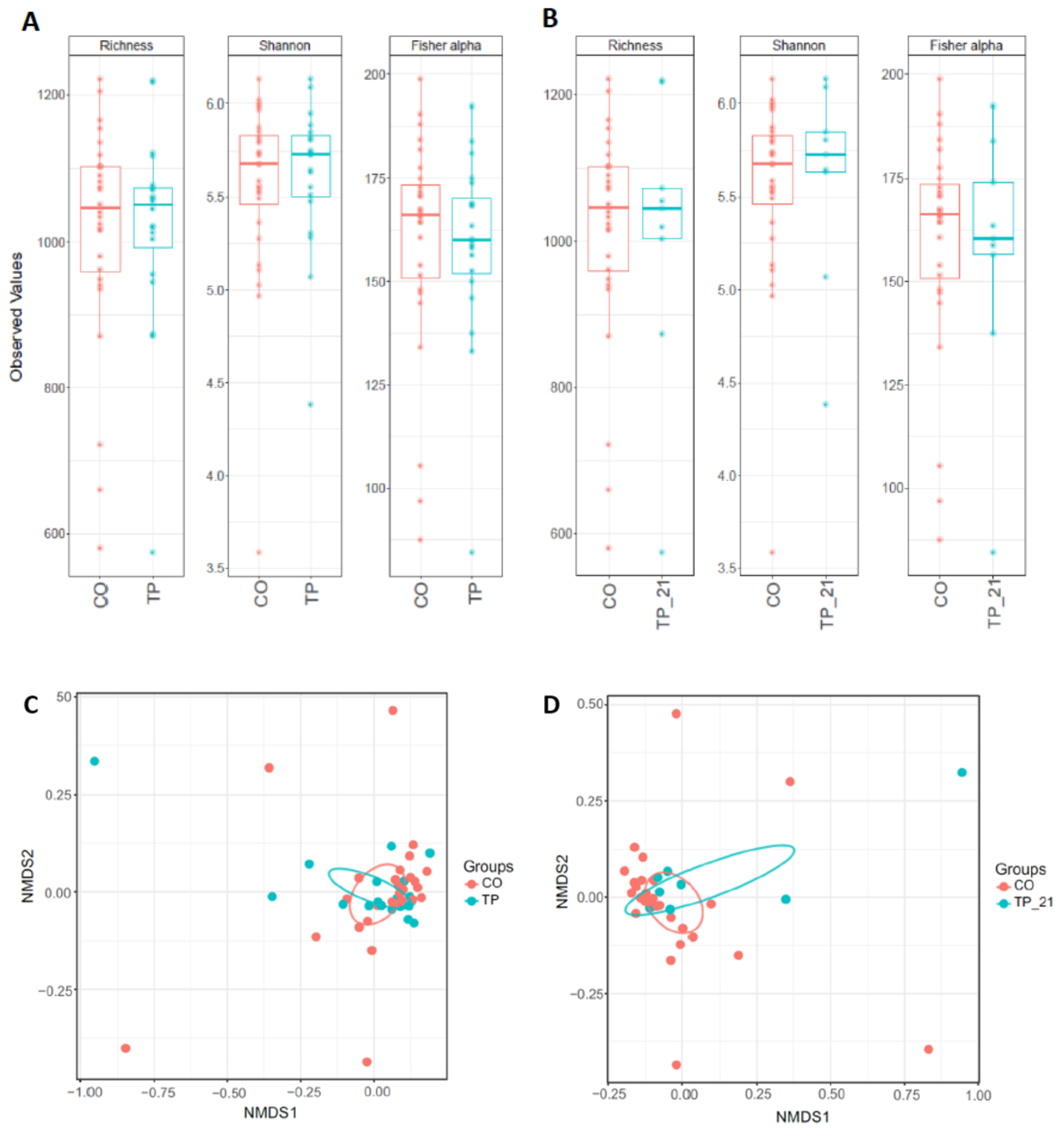
3.1. Microbiome Profiling
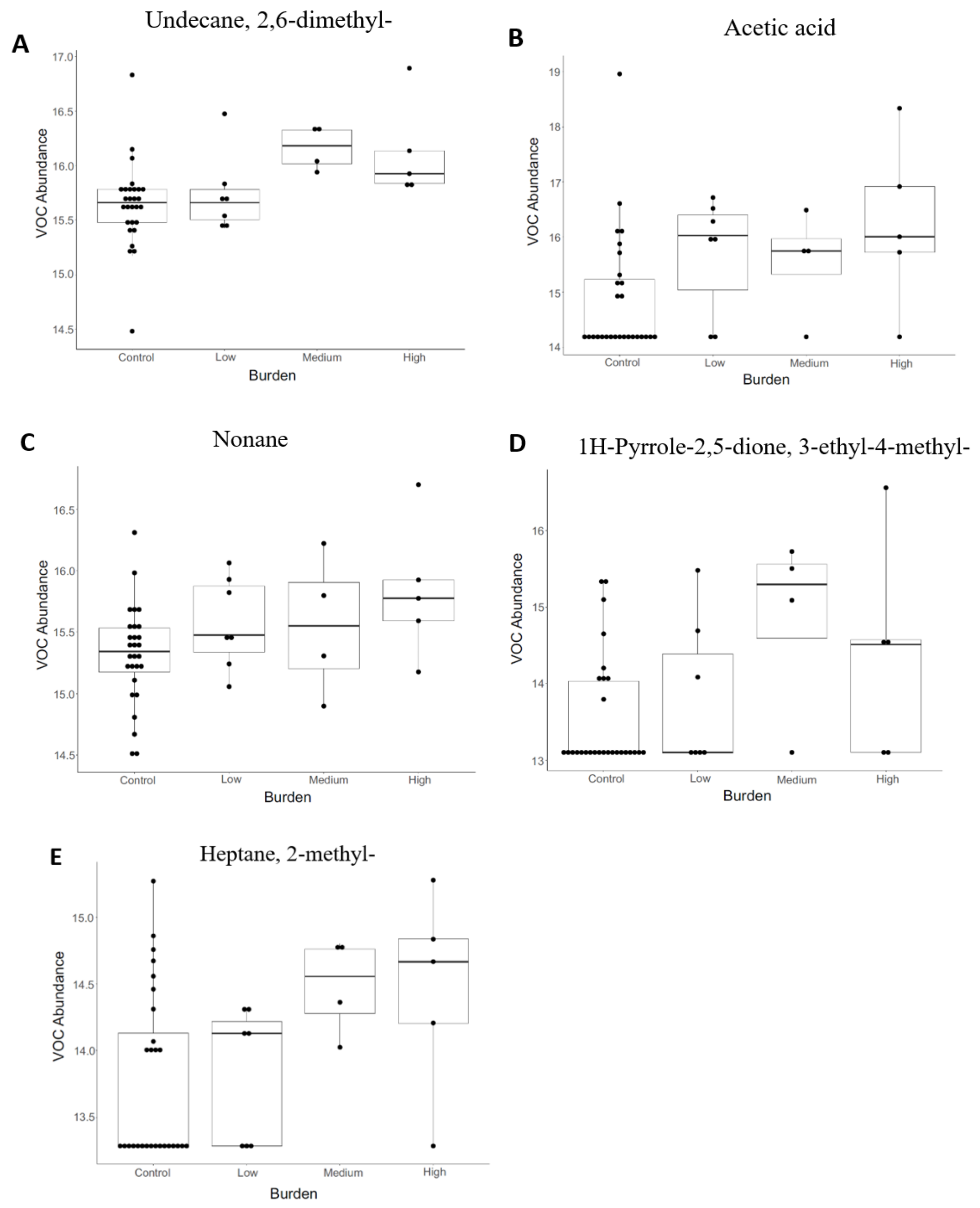
3.2. Volatile Organic Compound (VOC) Profiling
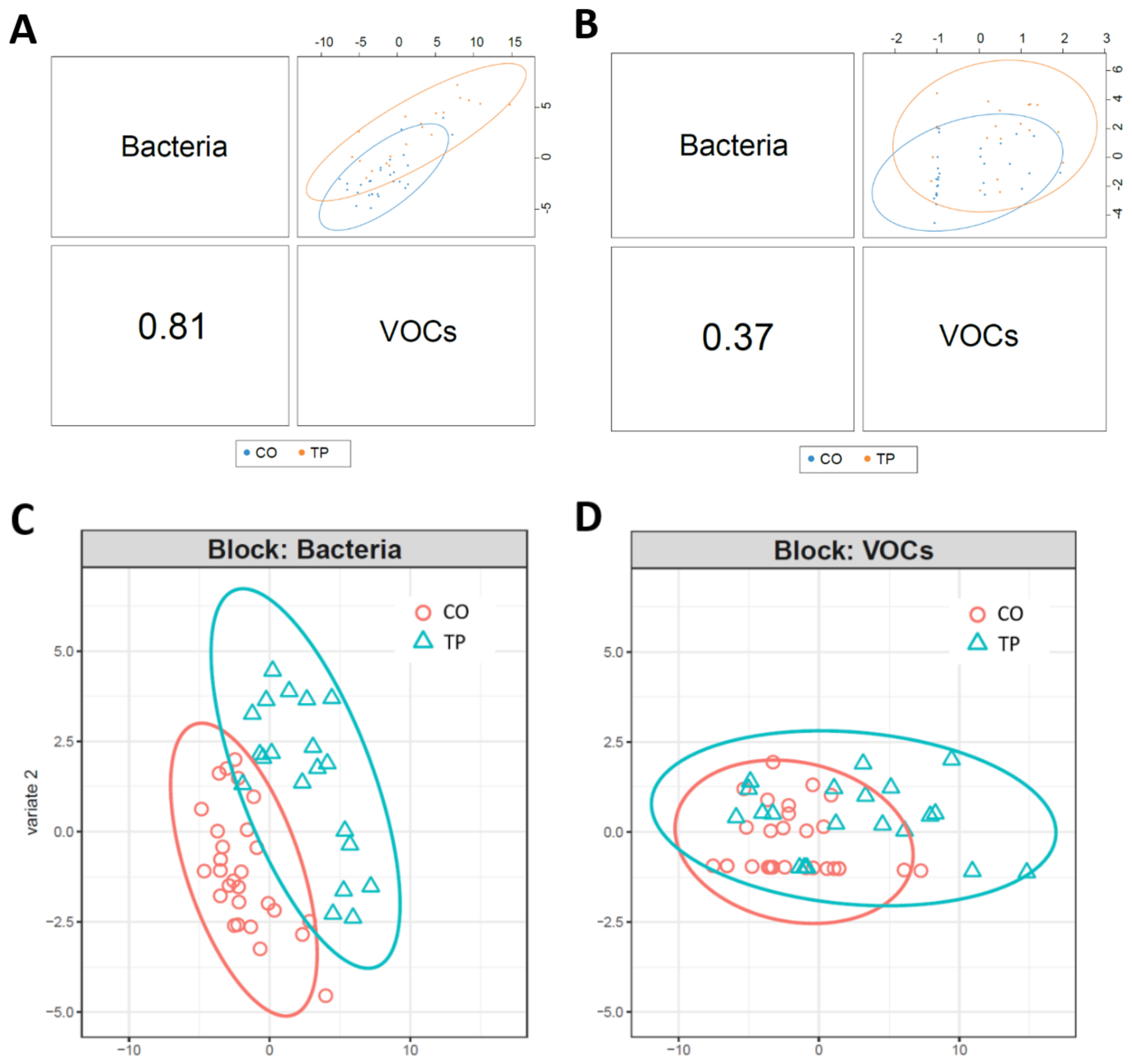
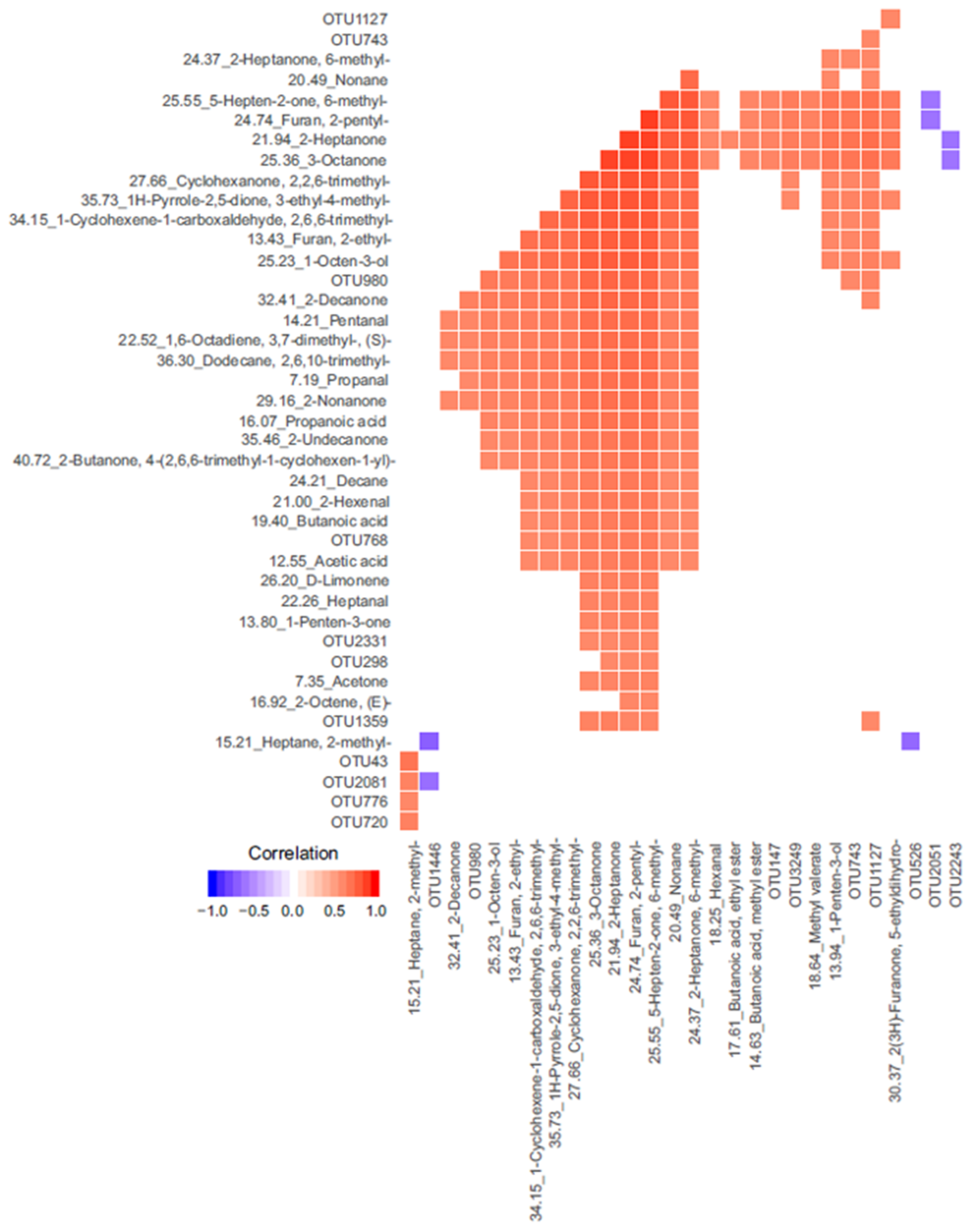
3.3. Integrated-Omics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomczuk, K.; Kostro, K.; Szczepaniak, K.O.; Grzybek, M.; Studzińska, M.; Demkowska-Kutrzepa, M.; Roczeń-Karczmarz, M. Comparison of the sensitivity of coprological methods in detecting Anoplocephala perfoliata invasions. Parasitol. Res. 2014, 113, 2401–2406. [Google Scholar] [CrossRef] [PubMed]
- Sallé, G.; Guillot, J.; Tapprest, J.; Foucher, N.; Sevin, C.; Laugier, C. Compilation of 29 years of postmortem examinations identifies major shifts in equine parasite prevalence from 2000 onwards. Int. J. Parasitol. 2020, 50, 125–132. [Google Scholar] [CrossRef]
- Owen, R.A.; Jagger, D.W.; Quan-Taylor, R. Caecal intussusceptions in horses and the significance of Anoplocephala perfoliata. Vet. Rec. 1989, 124, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Proudman, C.J.; French, N.P.; Trees, A.J. Tapeworm infection is a significant risk factor for spasmodic colic and ileal impaction colic in the horse. Equine Vet. J. 1998, 30, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Proudman, C.J.; Holdstock, N.B. Investigation of an outbreak of tapeworm-associated colic in a training yard. Equine Vet. J. 2000, 32, 37–41. [Google Scholar] [CrossRef]
- Ryu, S.H.; Bak, U.B.; Kim, J.G.; Yoon, H.J.; Seo, H.S.; Kim, J.T.; Park, J.Y.; Lee, C.W. Cecal rupture by Anoplocephala perfoliata infection in a thoroughbred horse in Seoul Race Park, South Korea. J. Vet. Sci. 2001, 2, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Boswinkel, M.; Sloet van Oldruitenborgh-Oosterbann, M. Correlation between colic and antibody levels against Anoplocephala perfoliata in horses in The Netherlands. Tijdschr. Diergeneeskd. 2007, 132, 508–512. [Google Scholar] [PubMed]
- Nielsen, M.; Reinemeyer, C.; Donecker, J.; Leathwick, D.; Marchiondo, A.; Kaplan, R. Anthelmintic resistance in equine parasites—Current evidence and knowledge gaps. Vet. Parasitol. 2014, 204, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.K. Equine tapeworm infections: Disease, diagnosis and control. Equine Vet. Educ. 2015, 28, 388–395. [Google Scholar] [CrossRef]
- Lyons, E.; Bellaw, J.; Dorton, A.; Tolliver, S. Efficacy of moxidectin and an ivermectin-praziquantel combination against ascarids, strongyles, and tapeworms in Thoroughbred yearlings in field tests on a farm in Central Kentucky in 2016. Vet. Parasitol. Reg. Stud. Rep. 2017, 8, 123–126. [Google Scholar] [CrossRef]
- Lightbody, K.L.; Davis, P.J.; Austin, C.J. Validation of a novel saliva-based ELISA test for diagnosing tapeworm burden in horses. Vet. Clin. Pathol. 2016, 45, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Proudman, C.; Trees, A. Use of excretory/secretory antigens for the serodiagnosis of Anoplocephala perfoliata cestodosis. Vet. Parasitol. 1996, 61, 239–247. [Google Scholar] [CrossRef]
- Abbott, J.B.; Mellor, D.J.; Barrett, E.J.; Proudman, C.J.; Love, S. Serological changes observed in horses infected with Anoplocephala perfoliata after treatment with praziquantel and natural reinfection. Vet. Rec. 2008, 162, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Proudman, C.; Trees, A. Correlation of antigen specific IgG and IgG(T) responses with Anoplocephala perfoliata infection intensity in the horse. Parasite Immunol. 1996, 18, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Proudman, C.; Edwards, G. Validation of a centrifugation/floatation technique for the diagnosis of equine cestodiasis. Vet. Rec. 1992, 131, 71–72. [Google Scholar] [CrossRef]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Daly, K.; Stewart, C.S.; Flint, H.J.; Shirazi-Beechey, S.P. Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes. FEMS Microbiol. Ecol. 2001, 38, 141–151. [Google Scholar] [CrossRef]
- Reynolds, L.A.; Smith, K.A.; Filbey, K.J.; Harcus, Y.; Hewitson, J.P.; Redpath, S.A.; Valdez, Y.; Yebra, M.J.; Finlay, B.B.; Maizels, R.M. Commensal-pathogen interactions in the intestinal tract: Lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes 2014, 5, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.M.; Harris, N.L. Interactions between the intestinal microbiome and helminth parasites. Parasite Immunol. 2016, 38, 5–11. [Google Scholar] [CrossRef]
- Brosschot, T.P.; Reynolds, L.A. The impact of a helminth-modified microbiome on host immunity. Mucosal Immunol. 2018, 11, 1039–1046. [Google Scholar] [CrossRef]
- Clark, A.; Sallé, G.; Ballan, V.; Reigner, F.; Meynadier, A.; Cortet, J.; Koch, C.; Riou, M.; Blanchard, A.; Mach, N. Strongyle Infection and Gut Microbiota: Profiling of Resistant and Susceptible Horses over a grazing season. Front. Physiol. 2018, 9, 272. [Google Scholar] [CrossRef] [PubMed]
- Peachey, L.; Molena, R.; Jenkins, T.; Di Cesare, A.; Traversa, D.; Hodgkinson, J.; Cantacessi, C. The relationships between faecal egg counts and gut microbial composition in UK Thoroughbreds infected by cyathostomins. Int. J. Parasitol. 2018, 48, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Peachey, L.E.; Castro, C.; Molena, R.A.; Jenkins, T.P.; Griffin, J.L.; Cantacessi, C. Dysbiosis associated with acute helminth infections in herbivorous youngstock—observations and implications. Sci. Rep. 2019, 9, 11121. [Google Scholar] [CrossRef]
- Walshe, N.; Duggan, V.; Cabrera-Rubio, R.; Crispie, F.; Cotter, P.; Feehan, O.; Mulcahy, G. Removal of adult cyathostomins alters faecal microbiota and promotes an inflammatory phenotype in horses. Int. J. Parasitol. 2019, 49, 489–500. [Google Scholar] [CrossRef]
- Daniels, S.P.; Leng, J.; Swann, J.R.; Proudman, C.J. Bugs and drugs: A systems biology approach to characterising the effect of moxidectin on the horse’s faecal microbiome. Anim. Microbiome 2020, 2, 1–14. [Google Scholar] [CrossRef]
- Korpi, A.; Järnberg, J.; Pasanen, A.-L. Microbial volatile organic compounds. Crit. Rev. Toxicol. 2009, 39, 139–193. [Google Scholar] [CrossRef]
- Proudman, C.; Hunter, J.O.; Darby, A.C.; Escalona, E.E.; Batty, C.; Turner, C. Characterisation of the faecal metabolome and microbiome of Thoroughbred racehorses. Equine Vet. J. 2014, 47, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Salem, S.E.; Hough, R.; Probert, C.; Maddox, T.W.; Antczak, P.; Ketley, J.M.; Williams, N.J.; Stoneham, S.J.; Archer, D.C. A longitudinal study of the faecal microbiome and metabolome of periparturient mares. PeerJ 2019, 7, e6687. [Google Scholar] [CrossRef] [PubMed]
- Walshe, N.; Mulcahy, G.; Hodgkinson, J.; Peachey, L. No Worm is an island; the influence of commensal gut microbiota on cyathostomin infections. Animals 2020, 10, 2309. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Silva, G.; Ramos, R.; Staempfli, H.; Arroyo, L.; Kim, P.; Weese, J. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. Vet. J. 2015, 205, 74–80. [Google Scholar] [CrossRef]
- Dougal, K.; Harris, P.A.; Edwards, A.; Pachebat, J.A.; Blackmore, T.M.; Worgan, H.J.; Newbold, C.J. A comparison of the microbiome and the metabolome of different regions of the equine hindgut. FEMS Microbiol. Ecol. 2012, 82, 642–652. [Google Scholar] [CrossRef]
- Pearson, G.R.; Davies, L.W.; White, A.L.; O’Brien, J.K. Pathological lesions associated with Anoplocephala perfoliata at the ileo-caecal junction of horses. Vet. Rec. 1993, 132, 179–182. [Google Scholar] [CrossRef]
- Christie, M.; Jackson, F. Specific identification of strongyle eggs in small samples of sheep faeces. Res. Vet. Sci. 1982, 32, 113–117. [Google Scholar] [CrossRef]
- Blackmore, T.M.; Dugdale, A.; Argo, C.M.; Curtis, G.; Pinloche, E.; Harris, P.A.; Worgan, H.J.; Girdwood, S.E.; Dougal, K.; Newbold, C.J.; et al. Strong stability and host specific bacterial community in faeces of ponies. PLoS ONE 2013, 8, e75079. [Google Scholar] [CrossRef][Green Version]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.T.; Berg-Lyons, D.; Caporaso, J.G.; Walters, W.A.; Knight, R.; Fierer, N. Examining the global distribution of dominant archaeal populations in soil. ISME J. 2010, 5, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Hough, R.; Archer, D.; Probert, C. A comparison of sample preparation methods for extracting volatile organic compounds (VOCs) from equine faeces using HS-SPME. Metabolomics 2018, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files. Available online: https://github.com/najoshi/sickle/releases/tag/v1.2 (accessed on 29 November 2020).
- Mahé, F.; Rognes, T.; Quince, C.; De Vargas, C.; Dunthorn, M. Swarm v2: Highly-scalable and high-resolution amplicon clustering. PeerJ 2015, 3, e1420. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A fexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Ijaz, U.Z.; Sivaloganathan, L.; McKenna, A.; Richmond, A.; Kelly, C.; Linton, M.; Stratakos, A.C.; Lavery, U.; Elmi, A.; Wren, B.W.; et al. Comprehensive longitudinal microbiome analysis of the chicken cecum reveals a shift from competitive to environmental drivers and a window of opportunity for campylobacter. Front. Microbiol. 2018, 9, 2452. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.; Kindt, R.; Legendre, P.; Michen, P.; O’Hara, R.; Simpson, G.; Solymos, P.; Stevens, M.; Wagner, H. Vegan: Ordination Methods, Diversity Analysis and Other Functions for Community and Vegetation Ecologists. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 29 November 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Aggio, R.; Villas-Bôas, S.G.; Ruggiero, K. Metab: An R package for high-throughput analysis of metabolomics data generated by GC-MS. Bioinformatics 2011, 27, 2316–2318. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Steelman, S.M.; Chowdhary, B.P.; Dowd, S.; Suchodolski, J.; Janečka, J.E. Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet. Res. 2012, 8, 231. [Google Scholar] [CrossRef]
- Weese, J.S.; Holcombe, S.J.; Embertson, R.M.; Kurtz, K.A.; Roessner, H.A.; Jalali, M.; Wismer, S.E. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Vet. J. 2014, 47, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.M.; Jenkins, T.P.; Latrofa, M.S.; Giannelli, A.; Papadopoulos, E.; De Carvalho, L.M.; Nolan, M.J.; Otranto, D.; Cantacessi, C. Helminth infections and gut microbiota—A feline perspective. Parasites Vectors 2016, 9, 625. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.; Li, W.; Sun, J.; Yu, P.; Baldwin, R.L.; Urban, J.F. The effect of helminth infection on the microbial composition and structure of the caprine abomasal microbiome. Sci. Rep. 2016, 6, 20606. [Google Scholar] [CrossRef] [PubMed]
- Šlapeta, J.; Dowd, S.E.; Alanazi, A.D.; Westman, M.E.; Brown, G.K. Differences in the faecal microbiome of non-diarrhoeic clinically healthy dogs and cats associated with Giardia duodenalis infection: Impact of hookworms and coccidia. Int. J. Parasitol. 2015, 45, 585–594. [Google Scholar] [CrossRef]
- Li, R.W.; Wu, S.; Li, W.; Huang, Y.; Gasbarre, L.C. Metagenome plasticity of the bovine abomasal microbiota in immune animals in response to Ostertagia Ostertagi infection. PLoS ONE 2011, 6, e24417. [Google Scholar] [CrossRef]
- Houlden, A.; Hayes, K.S.; Bancroft, A.J.; Worthington, J.J.; Wang, P.; Grencis, R.K.; Roberts, I.S. Chronic trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: Effects reversed by pathogen clearance. PLoS ONE 2015, 10, e0125945. [Google Scholar] [CrossRef]
- Jenkins, T.P.; Peachey, L.E.; Ajami, N.J.; Macdonald, A.S.; Hsieh, M.H.; Brindley, P.J.; Cantacessi, C.; Rinaldi, G. Schistosoma mansoni infection is associated with quantitative and qualitative modifications of the mammalian intestinal microbiota. Sci. Rep. 2018, 8, 12072. [Google Scholar] [CrossRef]
- Cattadori, I.M.; Sebastian, A.; Hao, H.; Katani, R.; Albert, I.; Eilertson, K.E.; Kapur, V.; Pathak, A.; Mitchell, S. Impact of helminth infections and nutritional constraints on the small intestine microbiota. PLoS ONE 2016, 11, e0159770. [Google Scholar] [CrossRef]
- Arrazuria, R.; Elguezabal, N.; Juste, R.A.; Derakhshani, H.; Khafipour, E. Mycobacterium avium subspecies paratuberculosis infection modifies gut microbiota under different dietary conditions in a rabbit model. Front. Microbiol. 2016, 7, 446. [Google Scholar] [CrossRef]
- Daly, K.; Proudman, C.J.; Duncan, S.H.; Flint, H.J.; Dyer, J.; Shirazi-Beechey, S.P. Alterations in microbiota and fermentation products in equine large intestine in response to dietary variation and intestinal disease. Br. J. Nutr. 2011, 107, 989–995. [Google Scholar] [CrossRef]
- Biddle, A.S.; Stewart, L.; Blanchard, J.L.; Leschine, S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Wu, S.; Li, R.W.; Li, W.; Beshah, E.; Dawson, H.D.; Urban, J.F. Worm burden-dependent disruption of the porcine colon microbiota by Trichuris suis infection. PLoS ONE 2012, 7, e35470. [Google Scholar] [CrossRef]
- Nagpal, D.; Prakash, S.; Bhat, K.; Singh, G. Detection and comparison of Selenomonas sputigena in subgingival biofilms in chronic and aggresive periodontitis patients. Indian Soc. Periodontol. 2016, 20, 286–291. [Google Scholar]
- Shirazi-Beechey, S.P. Molecular insights into dietary induced colic in the horse. Equine Vet. J. 2008, 40, 414–421. [Google Scholar] [CrossRef]
- Williamson, R.; Gasser, R.; Middleton, D.; Beveridge, I. The distribution of Anoplocephala perfoliata in the intestine of the horse and associated pathological changes. Vet. Parasitol. 1997, 73, 225–241. [Google Scholar] [CrossRef]
- Barrett, E.J.; Blair, C.W.; Farlam, J.; Proudman, C.J. Postdosing colic and diarrhoea in horses with serological evidence of tapeworm infection. Vet. Rec. 2005, 156, 252–253. [Google Scholar] [CrossRef] [PubMed]
- Dougal, K.; De La Fuente, G.; Harris, P.A.; Girdwood, S.E.; Pinloche, E.; Geor, R.J.; Nielsen, B.D.; Schott, H.C.; Elzinga, S.; Newbold, C.J. Characterisation of the faecal bacterial community in adult and elderly horses fed a high fibre, high oil or high starch diet using 454 pyrosequencing. PLoS ONE 2014, 9, e87424. [Google Scholar] [CrossRef]
- Yamano, H.; Koike, S.; Kobayashi, Y.; Hata, H. Phylogenetic analysis of hindgut microbiota in Hokkaido native horses compared to light horses. Anim. Sci. J. 2008, 79, 234–242. [Google Scholar] [CrossRef]
- Costa, M.C.; Stämpfli, H.R.; Arroyo, L.G.; Allen-Vercoe, E.; Gomes, R.G.; Weese, J.S. Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs. BMC Vet. Res. 2015, 11, 19. [Google Scholar] [CrossRef]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- De Raad, M.; Fischer, C.R.; Northen, T.R. High-throughput platforms for metabolomics. Curr. Opin. Chem. Biol. 2016, 30, 7–13. [Google Scholar] [CrossRef]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis—a marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, S.; Bobeldijk, I.; Verheij, E.R.; Ramaker, R.; Kochhar, S.; Macdonald, I.A.; Van Ommen, B.; Smilde, A.K. Large-scale human metabolomics studies: A strategy for data (pre-) processing and validation. Anal. Chem. 2006, 78, 567–574. [Google Scholar] [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Effmert, U.; Kalderás, J.; Warnke, R.; Piechulla, B. Volatile mediated interactions between bacteria and fungi in the soil. J. Chem. Ecol. 2012, 38, 665–703. [Google Scholar] [CrossRef] [PubMed]
- Hertel, M.; Hartwig, S.; Schütte, E.; Gillissen, B.; Preissner, R.; Schmidt-Westhausen, A.M.; Paris, S.; Kastner, I.; Preissner, S. Identification of signature volatiles to discriminate Candida albicans, glabrata, krusei and tropicalis using gas chromatography and mass spectrometry. Mycoses 2016, 59, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019. [Google Scholar] [CrossRef]
- Nielsen, M.; Baptiste, K.; Tolliver, S.; Collins, S.; Lyons, E. Analysis of multiyear studies in horses in Kentucky to ascertain whether counts of eggs and larvae per gram of feces are reliable indicators of numbers of strongyles and ascarids present. Vet. Parasitol. 2010, 174, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Dowdall, S.M.J.; Matthews, J.B.; Mair, T.; Murphy, D.; Love, S.; Proudman, C.J. Antigen-specific IgG(T) responses in natural and experimental cyathostominae infection in horses. Vet. Parasitol. 2002, 106, 225–242. [Google Scholar] [CrossRef]
- Tzelos, T.; Geyer, K.K.; Mitchell, M.C.; McWilliam, H.E.G.; Kharchenko, V.O.; Burgess, S.T.G.; Matthews, J.B. Characterisation of serum IgG(T) responses to potential diagnostic antigens for equine cyathostominosis. Int. J. Parasitol. 2020, 50, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Peachey, L.E.; Jenkins, T.P.; Cantacessi, C. This gut ain’t big enough for both of us. Or is it? Helminth–microbiota interactions in veterinary species. Trends Parasitol. 2017, 33, 619–632. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | p-Value (FDR Corrected) | More Abundant in |
|---|---|---|
| TP vs. CO | ||
| Family | ||
| Bacteroidales UCG-001 | 0.016 | CO |
| Genus | ||
| Ruminococcaceae UCG-004 | 0.006 | CO |
| Jeotgalicoccus | 0.012 | TP |
| Candidatus Soleaferrea | 0.012 | CO |
| Romboutsia | 0.028 | CO |
| TP_21 vs. CO | p-value (FDR corrected) | More abundant in |
| Order | ||
| Rickettsiales | 0.004 | CO |
| Family | ||
| Rickettsiales Incertae Sedis | 0.005 | CO |
| Genus | ||
| Candidatus Hepatincola | 0.01 | CO |
| Selenomonas 3 | 0.02 | TP_21 |
| High_FEC vs. Low_FEC | p-value (FDR corrected) | More abundant in |
| Order | ||
| Rhodospirillales | 0.004 | High_FEC |
| Family | ||
| Fusobacteriaceae | 0.003 | High_FEC |
| Rhodospirillaceae | 0.003 | High_FEC |
| Bacteroidaceae | 0.003 | High_FEC |
| Genus | ||
| Bacteroides | 0.005 | High_FEC |
| Fusobacterium | 0.006 | High_FEC |
| Thalassospira | 0.007 | High_FEC |
| Gordonibacter | 0.021 | High_FEC |
| Prevotellaceae Ga6A1 group | 0.031 | High_FEC |
| TP vs. CO | p-Value | Adjusted p-Value | More Abundant in |
|---|---|---|---|
| VOCs | |||
| Undecane, 2,6-dimethyl- | 0.006 | 0.147 | TP |
| Acetic acid | 0.006 | 0.147 | TP |
| Nonane | 0.008 | 0.147 | TP |
| Furan, 2-methyl- | 0.009 | 0.147 | TP |
| 2-Undecanone | 0.013 | 0.147 | TP |
| 1H-Pyrrole-2,5-dione, 3-ethyl-4-methyl- | 0.013 | 0.147 | TP |
| Dodecane, 2,6,10-trimethyl- | 0.014 | 0.147 | TP |
| 3-Pentanone, 2-methyl- | 0.015 | 0.147 | TP |
| Heptane, 2-methyl- | 0.017 | 0.147 | TP |
| 2-Heptanone, 6-methyl- | 0.019 | 0.147 | TP |
| 2-Nonanone | 0.020 | 0.147 | TP |
| 2-Octene, (E)- | 0.025 | 0.147 | TP |
| 2-Decanone | 0.026 | 0.147 | TP |
| 2-Octanone | 0.026 | 0.147 | TP |
| 1-Penten-3-one | 0.026 | 0.147 | TP |
| Cyclohexanone, 2,2,6-trimethyl- | 0.029 | 0.147 | TP |
| 3-Octanone | 0.030 | 0.147 | TP |
| D-Limonene | 0.033 | 0.147 | TP |
| 2-Butanone | 0.033 | 0.147 | TP |
| Propanal | 0.035 | 0.147 | TP |
| Furan, 2-pentyl- | 0.036 | 0.147 | TP |
| 2-Heptanone | 0.038 | 0.147 | TP |
| 2-Butanone, 4-(2,6,6-trimethyl-1-cyclohexen-1-yl)- | 0.042 | 0.156 | TP |
| 1-Cyclohexene-1-carboxaldehyde, 2,6,6-trimethyl- | 0.044 | 0.156 | TP |
| TP_21 vs. CO | p-value | Adjusted p-value | More abundant in |
| VOCs | |||
| Undecane, 2,6-dimethyl- | 0.001 | 0.127 | TP_21 |
| 1H-Pyrrole-2,5-dione, 3-ethyl-4-methyl- | 0.005 | 0.128 | TP_21 |
| Heptane, 2-methyl- | 0.006 | 0.128 | TP_21 |
| 2-Heptanone, 6-methyl- | 0.006 | 0.128 | TP_21 |
| 1-Cyclohexene-1-carboxaldehyde, 2,6,6-trimethyl- | 0.012 | 0.174 | TP_21 |
| 2-Undecanone | 0.015 | 0.174 | TP_21 |
| Cyclohexanone, 2,2,6-trimethyl- | 0.015 | 0.174 | TP_21 |
| 2-Octene, (E)- | 0.017 | 0.174 | TP_21 |
| Furan, 2-pentyl- | 0.020 | 0.174 | TP_21 |
| Acetic acid | 0.021 | 0.174 | TP_21 |
| Octane | 0.023 | 0.178 | TP_21 |
| Nonane | 0.027 | 0.189 | TP_21 |
| Decane | 0.031 | 0.192 | TP_21 |
| Furan, 2-methyl- | 0.034 | 0.192 | TP_21 |
| TP_21 vs. CO | p-value | Adjusted p-value | More abundant in |
| 5-Hepten-2-one, 6-methyl- | 0.036 | 0.192 | TP_21 |
| 2-Decanone | 0.036 | 0.192 | TP_21 |
| 1-Octen-3-ol | 0.041 | 0.203 | TP_21 |
| High vs. Low FEC | |||
| VOCs | |||
| 1-Propanol, 2-methyl | 0.0219 | 0.903 | High_FEC |
| 1-Hexanol | 0.0381 | 0.903 | High_FEC |
| OTU | p-Value | OTU More Abundant in | VOCs Significantly Correlated | Correlation | VOC More Abundant in |
|---|---|---|---|---|---|
| OTU5074 | <0.001 | CO | Acetic acid | −0.42 | TP |
| OTU3878 | <0.001 | TP | 2-Nonanone | 0.46 | TP |
| 2-Decanone | 0.43 | TP | |||
| 1-Octen-3-ol | 0.43 | TP | |||
| OTU705 | <0.001 | CO | 2-Octanone | −0.46 | TP |
| 1-Octen-3-ol | −0.47 | TP | |||
| OTU314 | <0.001 | TP | 1H-Pyrrole-2,5-dione, 3-ethyl-4-methyl- | 0.43 | TP |
| Cyclohexanone, 2,2,6-trimethyl- | 0.41 | TP | |||
| Undecane, 2,6-dimethyl- | 0.41 | TP | |||
| 2-Heptanone, 6-methyl- | 0.4 | TP | |||
| 1-Cyclohexene-1-carboxaldehyde, 2,6,6-trimethyl- | 0.4 | TP | |||
| Dodecane, 2,6,10-trimethyl- | 0.37 | TP | |||
| Acetic acid | 0.37 | TP | |||
| OTU673 | <0.001 | CO | Heptane, 2-methyl- | −0.37 | TP |
| OTU2331 | <0.001 | TP | 2-Heptanone, 6-methyl- | 0.6 | TP |
| Decane | 0.58 | TP | |||
| Propanal | 0.56 | TP | |||
| 1-Cyclohexene-1-carboxaldehyde, 2,6,6-trimethyl- | 0.55 | TP | |||
| Furan, 2-pentyl- | 0.55 | TP | |||
| Cyclohexanone, 2,2,6-trimethyl- | 0.53 | TP | |||
| Undecane, 2,6-dimethyl- | 0.53 | TP | |||
| 1-Octen-3-ol | 0.51 | TP | |||
| 1H-Pyrrole-2,5-dione, 3-ethyl-4-methyl- | 0.48 | TP | |||
| 2-Heptanone | 0.45 | TP | |||
| Pentanal | 0.45 | TP | |||
| 2-Decanone | 0.43 | TP | |||
| Furan, 2-methyl- | 0.43 | TP | |||
| 3-Octanone | 0.42 | TP | |||
| 2-Nonanone | 0.4 | TP | |||
| 2-Undecanone | 0.4 | TP | |||
| Nonane | 0.37 | TP | |||
| OTU2051 | <0.001 | CO | Acetic acid | −0.37 | TP |
| OTU | p-value | OTU more abundant in | VOCs significantly correlated | Correlation | VOC more abundant in |
| 1H-Pyrrole-2,5-dione, 3-ethyl-4-methyl- | −0.38 | TP | |||
| 2-Heptanone | −0.39 | TP | |||
| 1-Octen-3-ol | −0.41 | TP | |||
| 2-Decanone | −0.45 | TP | |||
| 2-Octanone | −0.47 | TP | |||
| OTU142 | 0.015 | CO | 2-Nonanone | −0.37 | TP |
| Furan, 2-pentyl- | −0.38 | TP | |||
| 2-Butanone, 4-(2,6,6-trimethyl-1-cyclohexen-1-yl)- | −0.4 | TP | |||
| 2-Decanone | −0.43 | TP | |||
| Propanal | −0.43 | TP | |||
| 2-Heptanone | −0.43 | TP | |||
| 1-Octen-3-ol | −0.44 | TP | |||
| OTU147 | 0.015 | TP | 2-Heptanone | 0.56 | TP |
| 3-Octanone | 0.53 | TP | |||
| 1H-Pyrrole-2,5-dione, 3-ethyl-4-methyl- | 0.53 | TP | |||
| Furan, 2-pentyl- | 0.53 | TP | |||
| 2-Decanone | 0.51 | TP | |||
| 2-Nonanone | 0.51 | TP | |||
| 1-Penten-3-one | 0.49 | TP | |||
| Nonane | 0.48 | TP | |||
| 2-Heptanone, 6-methyl- | 0.47 | TP | |||
| 2-Undecanone | 0.47 | TP | |||
| 1-Octen-3-ol | 0.45 | TP | |||
| Cyclohexanone, 2,2,6-trimethyl- | 0.43 | TP | |||
| 2-Octanone | 0.43 | TP | |||
| 1-Cyclohexene-1-carboxaldehyde, 2,6,6-trimethyl- | 0.42 | TP | |||
| Propanal | 0.42 | TP | |||
| Furan, 2-methyl- | 0.41 | TP | |||
| Dodecane, 2,6,10-trimethyl- | 0.4 | TP | |||
| Acetic acid | 0.39 | TP | |||
| D-limonene | 0.36 | TP | |||
| OTU239 | 0.020 | CO | Decane | 0.41 | TP |
| 2-Heptanone, 6-methyl- | 0.39 | TP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slater, R.; Frau, A.; Hodgkinson, J.; Archer, D.; Probert, C. A Comparison of the Colonic Microbiome and Volatile Organic Compound Metabolome of Anoplocephala perfoliata Infected and Non-Infected Horses: A Pilot Study. Animals 2021, 11, 755. https://doi.org/10.3390/ani11030755
Slater R, Frau A, Hodgkinson J, Archer D, Probert C. A Comparison of the Colonic Microbiome and Volatile Organic Compound Metabolome of Anoplocephala perfoliata Infected and Non-Infected Horses: A Pilot Study. Animals. 2021; 11(3):755. https://doi.org/10.3390/ani11030755
Chicago/Turabian StyleSlater, Rachael, Alessandra Frau, Jane Hodgkinson, Debra Archer, and Chris Probert. 2021. "A Comparison of the Colonic Microbiome and Volatile Organic Compound Metabolome of Anoplocephala perfoliata Infected and Non-Infected Horses: A Pilot Study" Animals 11, no. 3: 755. https://doi.org/10.3390/ani11030755
APA StyleSlater, R., Frau, A., Hodgkinson, J., Archer, D., & Probert, C. (2021). A Comparison of the Colonic Microbiome and Volatile Organic Compound Metabolome of Anoplocephala perfoliata Infected and Non-Infected Horses: A Pilot Study. Animals, 11(3), 755. https://doi.org/10.3390/ani11030755