
Correlation of Genomic and Pedigree Inbreeding Coefficients in Small Cattle Populations

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Fped and Its Relation with Genomic Inbreeding Coefficients
3.2. Froh and the Genome Proportion with ROH
3.3. Froh with Different Lengths of ROH and Its Relationship with Other Inbreeding Coefficients
3.4. Fmg and Its Relationship with the Other Inbreeding Coefficients
3.5. Fgeno and the Possibility of Mating with Different Populations
3.6. Influence of the Origin Parents’ Country on The Level of Inbreeding in the Offspring
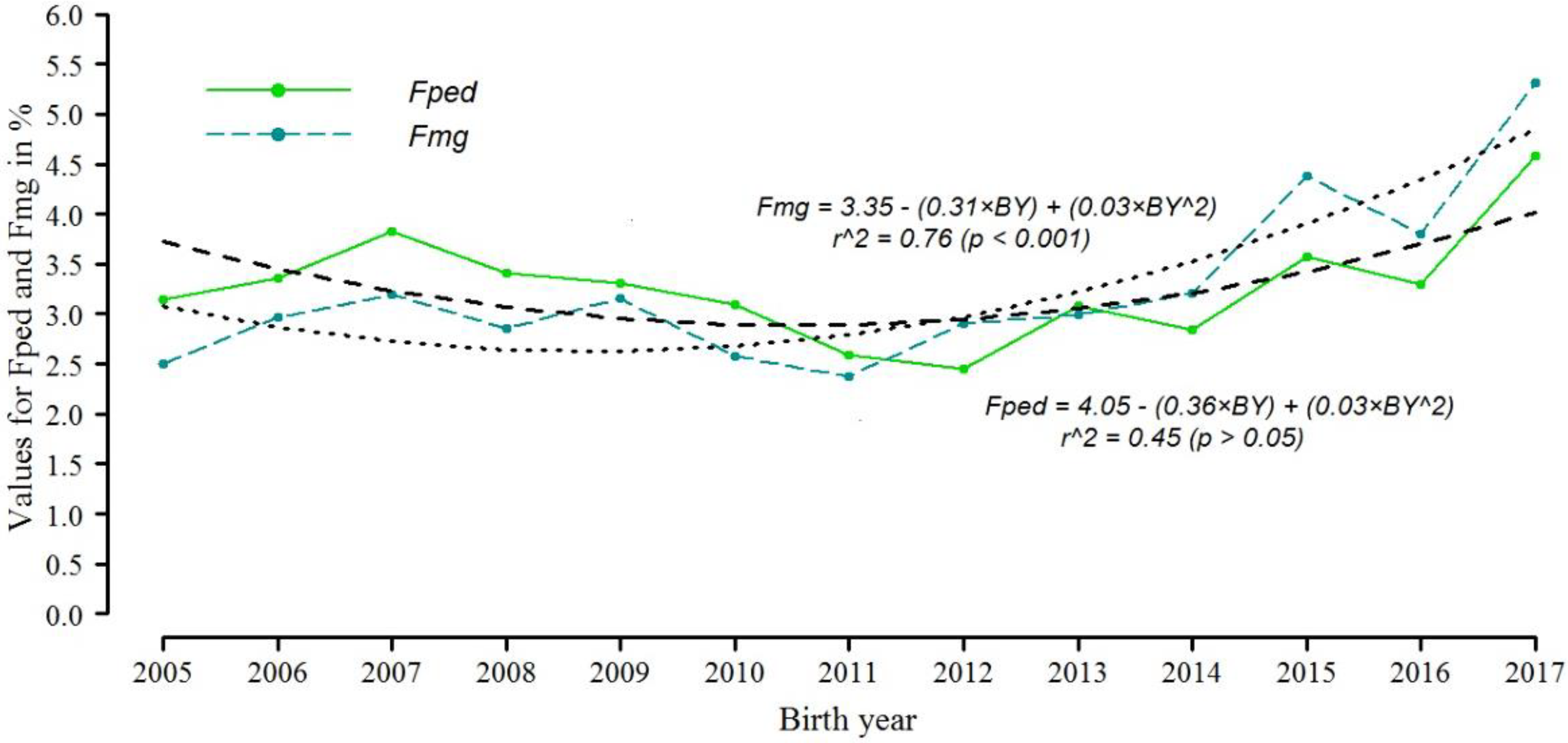
3.7. Inbreeding Coefficient Trends
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López-Gatius, F. Factors of a noninfectious nature affecting fertility after artificial insemination in lactating dairy cows. A review. Theriogenology 2012, 77, 1029–1041. [Google Scholar] [CrossRef]
- Wiggans, G.R.; Cole, J.B.; Hubbard, S.M.; Sonstegard, T.S. Genomic Selection in Dairy Cattle: The USDA Experience. Annu. Rev. Anim. Biosci. 2016, 5, 309–327. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ramilo, S.T.; Fernández, J.; Toro, M.A.; Hernández, D.; Villanueva, B. Genome-Wide estimates of coancestry, inbreeding and effective population size in the Spanish Holstein Population. PLoS ONE 2015, 10, e0124157. [Google Scholar] [CrossRef] [Green Version]
- Pryce, J.E.; Haile-mariam, M.; Goddard, M.E.; Hayes, B.J. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle. Genet. Sel. Evol. 2014, 46, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, S.; Tolone, M.; Gerlando RDi Fontanesi, L.; Sardina, M.T.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Anim. Consort. 2016, 10, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S. Coefficients of Inbreeding and Relationship. Am. Nat. 1922, 56, 330–338. Available online: https://www.jstor.org/stable/2456273 (accessed on 7 September 2021). [CrossRef] [Green Version]
- Guitou, H. Coeficiente de consanguinidad de un individuo. Sistemas de apareamiento. Rev. Angus 2010, 248, 31–37. [Google Scholar]
- Mcquillan, R.; Leutenegger, A.; Abdel-rahman, R.; Franklin, C.S.; Pericic, M.; Barac-lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Rebelato, A.B.; Caetano, A.R. Runs of homozygosity for autozygosity estimation and genomic analysis in production animals. Pesqui. Agropecuária Bras. 2018, 53, 975–984. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, I.; Misztal, I.; Legarra, A.; Tsuruta, S. Efficient computation of the genomic relationship matrix and other matrices used in single-step evaluation. J. Anim. Breed. Genet. 2011, 128, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; Mcparland, S.; Bradley, D.G. Runs of Homozygosity and Population History in Cattle. BMC Genet. 2012, 13, 2–11. Available online: http://www.biomedcentral.com/1471-2156/13/70%0ARESEARCH (accessed on 5 June 2021). [CrossRef] [Green Version]
- Kim, E.-S.; Sonstegard, T.S.; Van Tassell, C.P.; Wiggans, G.; Rothschild, M.F. The Relationship between Runs of Homozygosity and Inbreeding in Jersey Cattle under Selection. PLoS ONE 2015, 10, e0129967. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4496098 (accessed on 10 November 2021). [CrossRef] [PubMed] [Green Version]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.R.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef]
- Tier, B.; Meyr, K.M.F. Which Genomic Relationship Matrix? In Proceedings of the 21st Conference of the Association for the Advancement of Animal Breeding and Genetics, Lorne, Australia, 28–30 September 2015; pp. 28–30. Available online: https://hdl.handle.net/1959.11/19010 (accessed on 10 November 2021).
- Lozada-Soto, E.A.; Maltecca, C.; Lu, D.; Miller, S.; Cole, J.B.; Tiezzi, F. Trends in genetic diversity and the effect of inbreeding in American Angus cattle under genomic selection. Genet. Sel. Evol. 2021, 53, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Boichard, D. Pedig: A fortran package for pedigree analysis suited for large populations. In Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, 19–23 August 2002; Available online: https://hal.inrae.fr/hal-02833573/document (accessed on 10 November 2021).
- Aguilar, I.; Misztal, I. Technical note: Recursive algorithm for inbreeding coefficients assuming nonzero inbreeding of unknown parents. J. Dairy Sci. 2008, 91, 1669–1672. [Google Scholar] [CrossRef]
- VanRaden, P.M.; O’Connell, J.R.; Wiggans, G.R.; Weigel, K.A. Genomic evaluations with many more genotypes. Genet. Sel. Evol. 2011, 43, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2981575 (accessed on 10 November 2021). [CrossRef] [Green Version]
- Van Raden, P.M.; Olson, K.M.; Wiggans, G.R.; Cole, J.B.; Tooker, M.E. Genomic Inbreeding and Relationships among Holsteins, Jerseys, and Brown Swiss. J. Dairy Sci. 2011, 94, 5673–5682. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22032391 (accessed on 20 October 2021). [CrossRef]
- Bjelland, D.W.; Weigel, K.A.; Vukasinovic, N.; Nkrumah, J.D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. J. Dairy Sci. 2013, 96, 4697–4706. [Google Scholar] [CrossRef]
- Aguilar, I.; Misztal, I.; Tsuruta, S.; Legarra, A. PREGSF90–POSTGSF90: Computational Tools for the Implementation of Single-step Genomic Selection and Genome-wide. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production PREGSF90, Vancouver, BC, Canada, 2014; p. 3. Available online: https://www.researchgate.net/publication/264973897%0APREGSF90 (accessed on 10 November 2021).
- Golden Helix, I.; Bozeman, M. SNP & Variation SuiteTM; (Version 8) [Software]; Golden Helix, Inc.: Bozeman, MT, USA; p. 8. Available online: https://www.goldenhelix.com/products/SNP_Variation/index.html (accessed on 10 November 2021).
- Wayne, W.D. Bioestadística: Base Para el Análisis de las Ciencias de la Salud, 4th ed.; Wayne, W.D., Ed.; Editorial Limusa, S.A. De C.V.: Limusa, México, 2017; pp. 400–474. [Google Scholar]
- SAS Institute. 2019. Available online: https://www.sas.com/es_mx/industry/life-sciences/solution/real-world-evidence.html (accessed on 10 November 2021).
- Vargas Rodríguez, Y.M.; Eduardo, A.; Valdivia, O.; Rodríguez, G.I.V. Regresión polinómica, una competencia indispensable para el tratamiento de datos en Cinética Química. ContactoS 2015, 98, 25–35. Available online: https://apprimac.com/wp-content/uploads/2019/07/regresion_polinomica.pdf (accessed on 1 November 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.r-project.org/ (accessed on 1 November 2021).
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2014, 46, 110–121. [Google Scholar] [CrossRef]
- Zhang, Q.; Calus, M.P.L.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Forutan, M.; Ansari Mahyari, S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Szmatoła, T.; Gurgul, A.; Ropka-Molik, K.; Jasielczuk, I.; Zabek, T.; Bugno-Poniewierska, M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest Sci. 2016, 188, 2–80. [Google Scholar] [CrossRef]
- García-Ruiz, A.; Ruiz-Lopez, F.J.; Wiggans, G.R.; Van Tassell, C.P.; Montaldo, H.H. Effect of reference population size and available ancestor genotypes on imputation of Mexican Holstein genotypes. J. Dairy Sci. 2015, 98, 3478–3484. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.H.E.; Sonesson, A.K.; Gebregiwergis, G.; Woolliams, J.A. Management of Genetic Diversity in the Era of Genomics. Front. Genet. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Makanjuola, B.O.; Miglior, F.; Abdalla, E.A.; Maltecca, C.; Schenkel, F.S.; Baes, C.F. Effect of genomic selection on rate of inbreeding and coancestry and effective population size of Holstein and Jersey cattle populations. J. Dairy Sci. 2020, 103, 5183–5199. [Google Scholar] [CrossRef]
- Caballero, A.; Villanueva, B.; Druet, T. On the estimation of inbreeding depression using different measures of inbreeding from molecular markers. Evol. Appl. 2021, 14, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Szmatoła, T.; Topolski, P.; Jasielczuk, I.; Żukowski, K.; Bugno-Poniewierska, M. The use of runs of homozygosity for estimation of recent inbreeding in Holstein cattle. J. Appl. Genet. 2016, 57, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, B.; Fernández, A.; Saura, M.; Caballero, A.; Fernández, J.; Morales-González, E.; Toro, M.A.; Pong-Wong, R. The value of genomic relationship matrices to estimate levels of inbreeding. Genet. Sel. Evol. 2021, 53, 1–17. [Google Scholar] [CrossRef]
- Andere, C.I.; Rubio, N.; Rodriguez, E.; Casanova, D.; Aguilar, I. Inbreeding analysis of the population of Holstein dairy cattle registered in the Official Milk Control System of the Argentine Republic. Rev. Investig. Agropecu. 2017, 43, 92–97. [Google Scholar]
- García-Ruiz, A.; Cole, J.B.; VanRaden, P.M.; Wiggans, G.R.; Ruiz-López, F.J.; Van Tassell, C.P. Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc. Natl. Acad. Sci. USA 2016, 113, E3995–E4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| F% | Fped 3.11 ± 2.30 | Fgeno −0.02 ± 3.55 | Froh 2.77 ± 0.71 | Fmg 3.03 ± 3.05 |
|---|---|---|---|---|
| Fped | 1 | |||
| Fgeno | 0.39 ± 0.013 | 1 | ||
| Froh | 0.30 ± 0.014 | 0.82 ± 0.005 | 1 | |
| Fmg | 0.41 ± 0.013 | 0.96 ± 0.001 | 0.82 ± 0.005 | 1 |
| Length Mb | No. ROH | % |
|---|---|---|
| 1–2 | 215,175 | 51.32 |
| 2–4 | 93,596 | 22.32 |
| 4–8 | 61,329 | 14.63 |
| 8–16 | 34,375 | 8.20 |
| >16 | 14,812 | 3.53 |
| Froh with Different Lengths of ROH | |||||
|---|---|---|---|---|---|
| 1–2 Mb (Froh 1–2) | 2–4 Mb (Froh 2–4) | 4–8 Mb (Froh 4–8) | 8–16 Mb (Froh 8–16) | >16 Mb (Froh >16) | |
| N° of animals | 3759 | ||||
| Fgeno | −0.01 ± 0.016 | 0.14 ± 0.016 * | 0.30 ± 0.015 * | 0.46 ± 0.013 * | 0.71 ± 0.008 * |
| Fped | −0.03 ± 0.016 | −0.02 ± 0.016 | 0.05 ± 0.016 | 0.15 ± 0.016 * | 0.34 ± 0.014 * |
| Fmg | 0.01 ± 0.016 | 0.16 ± 0.016 * | 0.30 ± 0.015 * | 0.46 ± 0.013 * | 0.69 ± 0.008 * |
| Sire’s Country of Origin | No of Calves | Fgeno | Fped | Froh | Fmg |
|---|---|---|---|---|---|
| MEX | 405 | 3.35 ± 0.19 | 4.87 ± 0.16 | 3.07 ± 0.04 | 5.60 ± 0.19 |
| USA | 1095 | 2.69 ± 0.08 | 3.33 ± 0.08 | 3.31 ± 0.02 | 5.31 ± 0.07 |
| CAN | 282 | 2.95 ± 0.16 | 3.48 ± 0.15 | 3.40 ± 0.04 | 5.55 ± 0.15 |
| NLD | 67 | 2.22 ± 0.23 | 3.55 ± 0.23 | 3.18 ± 0.06 | 4.99 ± 0.21 |
| Dam’s country of origin | |||||
| MEX | 1599 | 2.85 ± 0.07 | 3.68 ± 0.07 | 3.26 ± 0.02 | 5.38 ± 0.07 |
| USA | 32 | 2.33 ± 0.34 | 4.36 ± 0.38 | 3.17 ± 0.10 | 5.13 ± 0.29 |
| CAN | 23 | 3.11 ± 0.53 | 3.39 ± 0.48 | 3.19 ± 0.14 | 5.85 ± 0.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes-Hernández, J.; García-Ruiz, A.; Vásquez-Peláez, C.G.; Ruiz-Lopez, F.d.J. Correlation of Genomic and Pedigree Inbreeding Coefficients in Small Cattle Populations. Animals 2021, 11, 3234. https://doi.org/10.3390/ani11113234
Cortes-Hernández J, García-Ruiz A, Vásquez-Peláez CG, Ruiz-Lopez FdJ. Correlation of Genomic and Pedigree Inbreeding Coefficients in Small Cattle Populations. Animals. 2021; 11(11):3234. https://doi.org/10.3390/ani11113234
Chicago/Turabian StyleCortes-Hernández, José, Adriana García-Ruiz, Carlos Gustavo Vásquez-Peláez, and Felipe de Jesus Ruiz-Lopez. 2021. "Correlation of Genomic and Pedigree Inbreeding Coefficients in Small Cattle Populations" Animals 11, no. 11: 3234. https://doi.org/10.3390/ani11113234
APA StyleCortes-Hernández, J., García-Ruiz, A., Vásquez-Peláez, C. G., & Ruiz-Lopez, F. d. J. (2021). Correlation of Genomic and Pedigree Inbreeding Coefficients in Small Cattle Populations. Animals, 11(11), 3234. https://doi.org/10.3390/ani11113234