
Composition of Fecal Microbiota in Grazing and Feedlot Angus Beef Cattle


1
College of Animal Science, Xichang University, Xichang 615000, China
2
Farm Animal Genetic Resources Exploration and Innovation Key Laboratory of Sichuan Province, Sichuan Agricultural University, Chengdu 611130, China
*
Author to whom correspondence should be addressed.
Animals 2021, 11(11), 3167; https://doi.org/10.3390/ani11113167
Submission received: 26 September 2021
/
Revised: 3 November 2021
/
Accepted: 3 November 2021
/
Published: 5 November 2021
(This article belongs to the Section Cattle)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
This study is to investigate the difference of bovine fecal microbiota between grazing and feedlot Angus cattle. The fecal bacterial community was analyzed by high-throughput sequencing of 16S rRNA gene from six Angus cattle grazed on grassland and six Angus cattle fed on a feedlot. A total of 775 OTUs were taxonomically assigned to bacterial 12 phyla, 19 classes, 25 orders, 54 families, 141 genera, and 145 species. The dominant phyla were Firmicutes and Bacteroidetes. There was similar species richness between grazing and feedlot Angus beef, while species diversity was higher in feedlot Angus beef. The relative abundance of Firmicutes, Cyanobacteria, Elusimicrobia and Patescibacteria was significantly different between grazing and feedlot Angus beef (p < 0.05). At the genus level, five microbiotas were significantly different microbiotas between the two groups and all belonged to the Firmicutes phylum. These significant differences in microbiota composition between grazing and feedlot Angus beef may have an impact on the meat quality of Angus beef.
Abstract
This study is to investigate the difference in bovine fecal microbiota between grazing and feedlot Angus cattle. Fecal samples were collected from six Angus cattle grazed on grassland and six Angus cattle fed on a feedlot. The fecal bacterial community was analyzed by high-throughput sequencing of 16S rRNA gene. Sequencing of the V3–V4 region totally produced 1,113,170 effective tages that were computationally clustered into 775 operational taxonomic units (OTUs). These 775 OTUs were taxonomically assigned to bacterial 12 phyla, 19 classes, 25 orders, 54 families, 141 genera, and 145 species. The dominant phyla were Firmicutes and Bacteroidetes. There was similar species richness between grazing and feedlot Angus beef, while higher species diversity was observed in feedlot Angus beef. The relative abundance of Firmicutes, Cyanobacteria, Elusimicrobia and Patescibacteria was significantly different between grazing and feedlot Angus beef (p < 0.05). At a genus level, five microbiotas were significantly different between the two groups and all belonged to the Firmicutes phylum. These significant differences in microbiota composition between grazing and feedlot Angus beef may have an impact on the meat quality of Angus beef.
1. Introduction
Grazing-fed cattle are free-ranged on pastures with grass as the main feed, while feedlot-fed cattle are raised in feedlots with grains as their main feed. The grass is low in energy but high in fiber, on the contrary, grains are high in energy and easy to digest. Thus, there are significant differences between the two feeding methods in digestibility, growth rate and meat quality. Compared to feedlot-fed beef, grazing-fed beef has lower fatty acid content and higher vitamin content [1]. Because nowadays people are concerned about energy and nutritional balance, consumers are increasingly favoring grazing-fed beef [2]. The proportion of grazing-fed cattle breeding has been gradually increasing as a result of this consumer preference.
Cattle gut microbes interact with their host and participate in the host’s physiological activities such as food digestion, energy metabolism and nutrient absorption, which are closely related to the healthy growth state and methane emissions of the host. Cattle gut microbes are affected by various factors, among which the composition and nutrition of the diet is a key factor in shaping its community structure, abundance and activity. Firmicutes, Bacteroidetes, Proteobacteria, TM7 and Actinobacteria were the main dominant phylum in cattle feces, and their abundance changed significantly with different diets [3]. Firmicutes was the first dominant phylum followed by Bacteroidetes, Proteobacteria, TM7 and Actinobacteria in moderate grain and high grain diets, while Firmicutes was the first dominant phylum followed by TM7, Actinobacteria, Proteobacteria and Bacteroidetes in silage/forage diets [4]. The fecal Firmicute:Bacteroidetes ratio was smaller when beef were fed more than 10% of dietary distiller grain compared with that of a corn diet [5]. There is a tendency to a greater relative abundance of Bacteroidetes but lesser Firmicutes in fecal matter after adding antibiotics or dried yeast to the beef diet [6]. Firmicutes, Bacteroidetes, and Verrucomicrobia were also the dominant phyla in yak, and the Firmicute:Bacteroidetes ratio was significantly decreasing from winter grassland to feedlot feeding [7]. In dairy cattle, diet was the most important parameter to explain fecal microbiota richness, and fecal microbiota of mixed (forage and concentrate) diet was more richness than that of dry forage diet [8].
Angus cattle is well known for its meat quality around the world, and grazing-fed Angus is more favored by consumers. The gut microbiota is significantly different between the grazing- and feedlot-fed beef. This study focuses on understanding the fecal microbiota in grazing- and feedlot-fed beef cattle by 16S rRNA sequencing, and searching for key bacterial differences. This investigation is expected to provide useful information to develop suitable nutritional and management strategies for grazing-fed and feedlot-fed Angus cattle.
2. Material and Methods
2.1. Fecal Sample Collection
Twelve 14-month-old healthy male Angus cattle (body weight: 405.52 + 8.30 kg) were selected from two nearby farms located in the Liangshan Yi autonomous of Sichuan (China). Six cattle (ANG1~6, group F) were randomly selected from a herd, which was kept in one indoor feedlot and fed with the total mixed ration diet based on corn silage according to the National Research Council (NRC). Another six cattle (ANG7~12, group G) were selected from a herd which grazed in a wild grassland and supplemented only with mineral salt. All cattle were adapted to their current state for more than 3 months before fecal sampling. Samples were collected directly from the rectum of each cattle using a disposable glove, and were immediately frozen in liquid nitrogen in 2 mL cryotubes, and then stored at −80 °C until DNA extraction.
2.2. DNA Extraction and Sequencing
Genomic DNA was extracted from the fecal sample using a QIAamp DNA stool Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. The variable regions V3–V4 of the 16S rDNA gene were amplified by PCR (98 °C for 2 min, followed by 30 cycles of 98 °C for 30 s, 50 °C for 30 s, 72 °C for 1 min, and 72 °C for 5 min) using bacterial domain-specific primers (338F: 5′-ACTCCTACGGGAGGCAGCAG-3′ and 806R: 5′-GGACTACHVGGGTWTCTAAT-3′). The ends of the primers were added unique barcodes. The PCR products were detected by 1.8% agarose gel electrophoresis, and approximately 450 bp samples were chosen and purified, and built into sequencing libraries. Finally, pair-end sequencing was conducted on an Illumina HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA).
2.3. Bioinformatics Analysis
Raw tags were spliced by merging the overlapping regions between paired-end reads with FLASH V1.2.7 [9], and then they were quality-filtered under specific filtering conditions to obtain high-quality clean tags with Trimmomatic v0.33 [10]. To get effective tags, the chimera sequences were detected and removed by comparing tags with a reference database (Ribosomal Database Program) using UCHIME v4.2 [11]. The effective tags were grouped into operation taxonomic units (OTUs) at 97% sequence identity using Usearch software. The OTUs were annotated based on the taxonomic databases of Sliva (bacteria) and UNITE (fungi).
To get the species classification information of each OTU, we compared the representative sequence of OTU with the microbial reference database. Then we counted the community composition of each sample at each level (phylum, class, order, family, genus, species), and generated species abundance tables at different classification levels by QIIME software [12]. To understand the evolutionary relationship and abundance difference of microorganisms, the species abundance information is returned to the taxonomic system relationship tree of the database by using MEGAN software [13]. The alpha diversity were calculated by Mothur V.1.30 [14] and the beta diversity were analysed for different groups by QIIME for different groups. To analyze the functional differences, we inferred the composition of functional genes in the samples by PICRUSt software [15]. And t-test is carried out between different groups and the significance level was set at p < 0.05.
3. Results
3.1. Gene Sequencing Data Summary
The V3–V4 hypervariable regions of 16S rRNA gene were sequenced to fecal microbial communities of 12 Angus cattle. A total of 1,194,599 raw paired-end reads were generated from 12 samples (average: 99,550 ± 26,487, range: 60,156–145,482). And then 1,113,170 effective tages were obtained from 12 samples (average: 92,764 ± 24,560, range: 55,212–134,921) with an average of 411.75 bp per tag after the merging overlapping paired-reads, quality filtering and removing of chimeric sequences. According to the 97% sequence similarity, 775 OTUs were computationally constructed with 687.92 ± 21.48 (range: 659–721) as the mean number of OTUs per sample (Figure 1).
These 775 OTUs were taxonomically assigned to bacterial 12 phyla, 19 classes, 25 orders, 54 families, 141 genera, and 145 species.
3.2. Analysis of Bacterial Diversity
The alpha diversity (observed species and Shannon diversity index) was calculated to estimate species richness and diversity in the 12 fecal microbiota samples (Figure 2). There was no statistically significant difference between group F and group G (Figure 2A, p = 0.054 of Kruskal-W allis test) in observed species, and a significant difference between group F and group G (Figure 2B, p = 0.025 of Kruskal-W allis test) in Shannon diversity index, which indicated that there was a similar species richness between group F and group G, while a higher species diversity in group F.
Principal coordinated analysis (PCoA) based on the binary Jaccard and Bray Curtis methods of beta diversity were further used to analyse compositional differences in fecal microbiota between group F and group G. The animals clustered together according to their particular group, suggesting a compositional shift with respect to community membership and structure between different groups. Both Jaccard and Bray-Curtis distance-based PCoA showed a significant difference between group F and group G (Figure 3, ANOSIM, p < 0.05), which indicated that each group hosts its own distinct bacterial community.
3.3. Analysis of Bacterial Taxonomy and Function
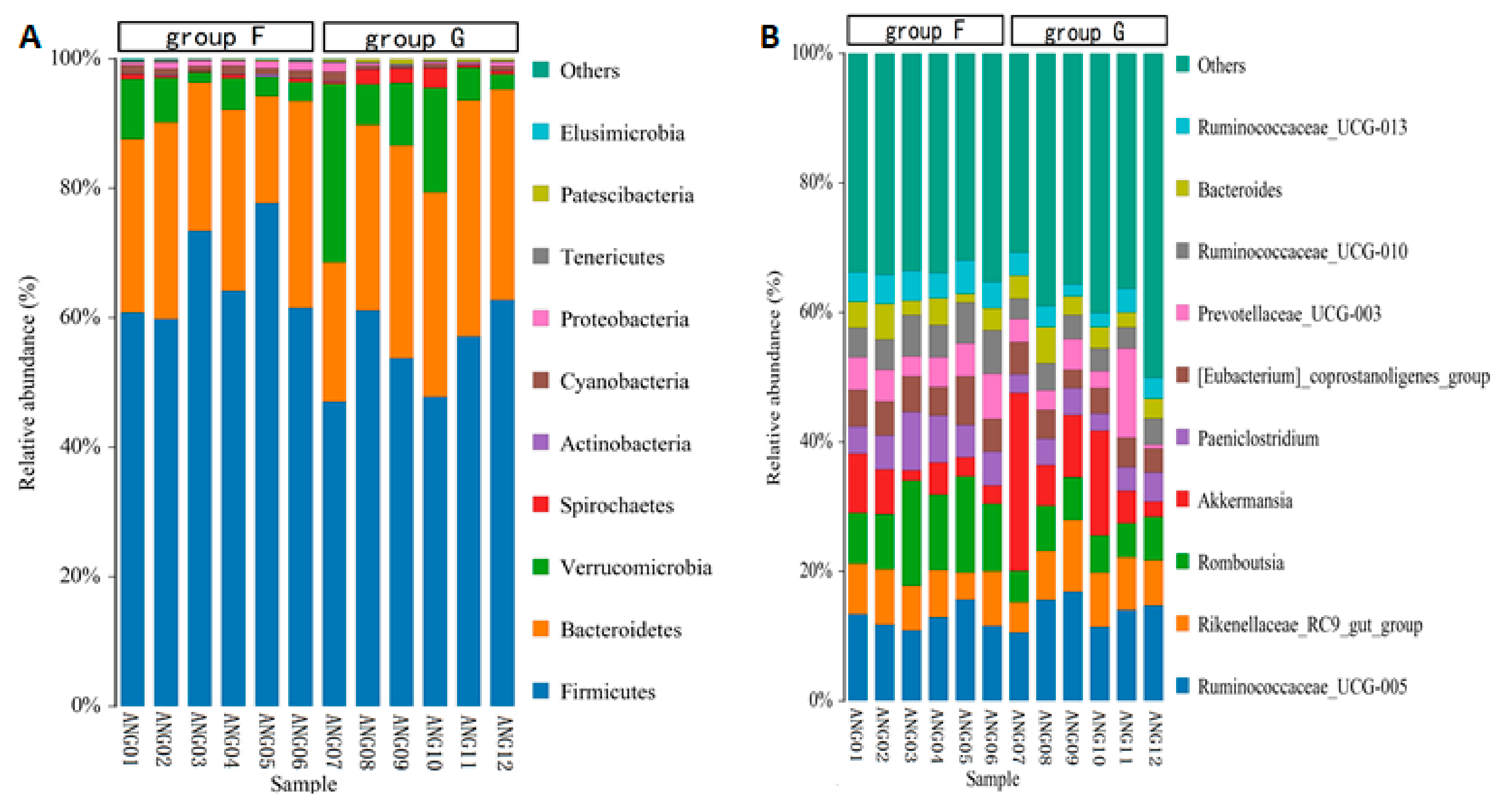
The taxonomic annotation at the phylum level showed that the common bacterial phyla of fecal microbiota in beef cattle were Firmicutes (with an average relative abundance of 60.61%), Bacteroidetes (28.32%), Verrucomicrobia (7.96%), Spirochaetes (0.91%), Cyanobacteria (0.84%), Proteobacteria (0.70%), Patescibacteria (0.25%), Tenericutes (0.22%), Actinobacteria (0.11%), and Elusimicrobia (0.04%), respectively (Figure 4A). The relative abundance of Firmicutes, Cyanobacteria and Elusimicrobia was significantly higher (p < 0.05) in group F compared with group G, while Patescibacteria was significantly lower (p < 0.05) in group F. At the genus level, the abundant genus was Ruminococcaceae_UCG-005 (13.30%), Romboutsia (8.82%), Akkermansia (7.96%), Rikenellaceae_RC9_gut_group (7.46%), Prevotellaceae_UCG-003 (4.83%), [Eubacterium]_coprostanoligenes_group (4.82%), Paeniclostridium (4.81%), Ruminococcaceae_UCG-010 (4.63%), Ruminococcaceae_UCG-013 (3.73%) and Bacteroides (3.44%), respectively (Figure 4B). The relative abundance of Romboutsia, Paeniclostridium, [Eubacterium]_coprostanoligenes_group, Ruminococcaceae_UCG-010 and Ruminococcaceae_UCG-013 was significantly higher (p < 0.05) in group F compared with group G.
4. Discussion
Grazing beef is more popular among consumers for its “natural” provenance and unique beef flavor. Both in dairy cow and beef, grazing was found to affect metabolic properties, thus affecting their skeletal muscle characteristics and content of nutritionally valuable compounds of meat [16]. Grazing Friesian bulls compared with tie-stall housed bulls, Type I %, Type IIA %, Type IIA and IIB fibre areas, capillarization, the activity of citrate synthase, glycogen content and pigmentation were higher in semitendinosus, longissimus dorsi, or supraspinatus muscles [17]. In Charolais steers, an increase mobility in pasture and a grass (vs. maize silage)-based diet contribute to the more oxidative metabolic orientation of muscles [18]. The gut microbiota of the farm animal is one of the important factors affecting the special flavor of meat [19]. In this study, we accordingly investigated the fecal microbiota composition between grazing and feedlot-fed cattle using high-throughput sequencing of 16S rRNA gene.
The composition proportion of fecal microbiota was different among individual, mainly due to diet, age, breed, veterinary drug, and geographic region. Several studies suggested that the community structure of cattle fecal microbiota was greatly affected by diet, especially between forage- and concentrate-based diets [20]. The cluster between forage- and concentrate-based cattle were separate from each other by fecal microbiota, and the difference among individuals of forage-based cattle is greater than that of concentrate-based cattle [4]. In this study, it shown a significant difference between group F and group G according to PCoA. The species diversity was higher in group F than that in group G. The diet composed of forage and grains to increase bacterial diversity in group F.
Firmicutes and Bacteroidetes were the main phyla, accounting for 90% of the cattle fecal bacteria. Firmicutes, the first dominant phylum in cattle fecal samples (>50%), was more abundant in concentrate-based diets than forage-based diets [4]. In this study, the abundance of Firmicutes was significantly different (p < 0.05) between the two diet groups with 66.26% in group F and 54.96% in group G. The five microbiotas were significantly different at the genus level between the two groups, all belonging to Firmicutes phylum. Firmicutes are beneficial bacteria in the intestinal tract, which help the host intestinal tract absorb energy from food [21]. The intestinal flora of obese mice has a strong ability to release energy from food, while the intestinal Firmicutes richness of obese mice is significantly higher than that of slim mice [22]. It is also reported that obese people have less Bacteroidetes than slim people, while there are more Firmicutes. After obese people lost weight through diet, obese peoples’ Bacteroidetes increased, while their Firmicutes counts decreased [23]. Here, not only were the Firmicutes of feedlot-fed cattle significantly higher than that of grazing-fed cattle, but also the Bacteroidetes of feedlot-fed cattle was lower than that of grazing-fed cattle (p > 0.05). Thus, the Firmicutes/Bacteroidetes ratio was higher in group F than that in group G. In human, the Firmicutes/Bacteroidetes ratio is used as a possible biomarker of gut dysbiosis [24].
Cyanobacteria, Patescibacteria and Elusimicrobia are less common microbes in cow feces, with an abundance of less than 1%. In this study, the relative abundance of Cyanobacteria, Patescibacteria, and Elusimicrobia was 0.84%, 0.25% and 0.04%, respectively. The relative abundance of Cyanobacteria and Elusimicrobia was significantly higher (p < 0.05) in group F compared with group G, while Patescibacteria was significantly lower (p < 0.05) in group F. Cyanobacteria was used as a source of functional food due to a potential natural alternative to antibiotics, antiviral or antifungal therapies [25]. The increased prevalence of Proteobacteria can be used as a marker for dysbiosis and a potential diagnostic criterion for disease in human gut [26]. Elusimicrobia are gut symbiotic anaerobic bacteria, which produce lactate, acetate, hydrogen and CO2 via fermentation [27]. The role of these microbiota in the cattle gut needs to be further studied.
Fecal microbiota transplantation (FMT) is an increasingly popular therapy for the treatment of diseases such as metabolic syndrome, diabetes, Crohn’s disease, Parkinson’s disease, multiple sclerosis, psoriasis, anorexia nervosa or Alzheimer disease in human [28]. Recently, FMT is successfully applied to ameliorate diarrhea and improve growth performance in pre-weaning calves [29]. In this study, the relative abundance of Firmicutes, Cyanobacteria, Elusimicrobia and Patescibacteria was significantly different between the feedlot- and grazing- Angus beef, which may provide the possibility to improve the quality of beef by FMT and microecologic agent.
In conclusion, the current study provides a view of fecal bacterial communities in grazing and feedlot Angus beef by 16S rRNA gene sequences. The 1,113,170 sequences were assigned to bacterial 12 phyla, 19 classes, 25 orders, 54 families, 141 genera, and 145 species. The common bacterial phyla of fecal microbiota were Firmicutes, Bacteroidetes, Verrucomicrobia, Spirochaetes, Cyanobacteria, Proteobacteria, Patescibacteria, Tenericutes, Actinobacteria and Elusimicrobia in order of abundance in Angus beef. There was similar species richness between grazing and feedlot Angus beef, while higher species diversity in feedlot Angus beef. The relative abundance of Firmicutes, Cyanobacteria and Elusimicrobia was significantly higher in feedlot Angus beef compared with grazing Angus beef, while Patescibacteria was significantly lower in feedlot Angus beef. At the genus level, five microbiotas were significantly different between the two groups, all belonging to the Firmicutes phylum. These significant differences in microbiota composition between grazing- and feedlot- Angus beef may have an impact on the meat quality of Angus beef.
Author Contributions
Conceptualization, X.J.; methodology, Z.Z. and L.Y.; software, Y.H.; validation, L.Y. and S.Z.; formal analysis, X.J.; investigation, Z.Z.; resources, Z.Z.; data curation, X.L.; writing—original draft preparation, Z.Z.; writing—review and editing, L.Y.; supervision, X.J.; project administration, X.J.; funding acquisition, Z.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Talent Introduction Program of Xichang University, grant number 50190007).
Institutional Review Board Statement
Collection of biological samples and experimental procedures involved in this study were approved by the Sichuan Agricultural University Animal Ethical and Welfare Committee, China (No. DKYB20200063).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Prache, S.; Martin, B.; Coppa, M. Review: Authentication of grass-fed meat and dairy products from cattle and sheep. Animal 2020, 14, 854–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, C.A.; Abbott, A.; Doyle, P.S.; A Nader, G.; Larson, S. A review of fatty acid profiles and antioxidant content in grass-fed and grain-fed beef. Nutr. J. 2010, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Wells, J.E. A Meta-analysis of Bacterial Diversity in the Feces of Cattle. Curr. Microbiol. 2015, 72, 145–151. [Google Scholar] [CrossRef]
- Kim, M.; Kim, J.; Kuehn, L.; Bono, J.L.; Berry, E.D.; Kalchayanand, N.; Freetly, H.C.; Benson, A.K.; Wells, J.E. Investigation of bacterial diversity in the feces of cattle fed different diets. J. Anim. Sci. 2014, 92, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Rice, W.C.; Galyean, M.L.; Cox, S.B.; Dowd, S.E.; Cole, N.A. Influence of wet distillers grains diets on beef cattle fecal bacterial community structure. BMC Microbiol. 2012, 12, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, T.; Jiao, P.; AlZahal, O.; Xie, X.; A Beauchemin, K.; Niu, D.; Yang, W. Fecal bacterial community of finishing beef steers fed ruminally protected and non-protected active dried yeast. J. Anim. Sci. 2020, 98, skaa058. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-L.; Xu, T.-W.; Wang, X.-G.; Geng, Y.-Y.; Liu, H.-J.; Hu, L.-Y.; Zhao, N.; Kang, S.-P.; Zhang, W.-M.; Xu, S.-X. The Effect of Transitioning between Feeding Methods on the Gut Microbiota Dynamics of Yaks on the Qinghai–Tibet Plateau. Animals 2020, 10, 1641. [Google Scholar] [CrossRef]
- Albonico, F.; Barelli, C.; Albanese, D.; Manica, M.; Partel, E.; Rosso, F.; Ripellino, S.; Pindo, M.; Donati, C.; Zecconi, A.; et al. Raw milk and fecal microbiota of commercial Alpine dairy cows varies with herd, fat content and diet. PLoS ONE 2020, 15, e0237262. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; I Gordon, J.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.-J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Mwangi, F.W.; Charmley, E.; Gardiner, C.P.; Malau-Aduli, B.S.; Kinobe, R.T.; Malau-Aduli, A.E.O. Diet and Genetics Influence Beef Cattle Performance and Meat Quality Characteristics. Foods 2019, 8, 648. [Google Scholar] [CrossRef] [Green Version]
- Vestergaard, M.; Oksbjerg, N.; Henckel, P. Influence of feeding intensity, grazing and finishing feeding on muscle fibre characteristics and meat colour of semitendinosus, longissimus dorsi and supraspinatus muscles of young bulls. Meat Sci. 1999, 54, 177–185. [Google Scholar] [CrossRef]
- Cassar-Malek, I.; Jurie, C.; Bernard, C.; Barnola, I.; Micol, D.; Hocquette, J.-F. Pasture-feeding of Charolais steers influences skeletal muscle metabolism and gene expression. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2009, 60, 83–90. [Google Scholar]
- Sun, J.; Wang, Y.; Li, N.; Zhong, H.; Xu, H.; Zhu, Q.; Liu, Y. Comparative Analysis of the Gut Microbial Composition and Meat Flavor of Two Chicken Breeds in Different Rearing Patterns. BioMed. Res. Int. 2018, 2018, 4343196. [Google Scholar] [CrossRef] [Green Version]
- Durso, L.M.; Harhay, G.; Smith, T.P.L.; Bono, J.L.; DeSantis, T.Z.; Harhay, D.M.; Andersen, G.; Keen, J.E.; Laegreid, W.W.; Clawson, M.L. Animal-to-Animal Variation in Fecal Microbial Diversity among Beef Cattle. Appl. Environ. Microbiol. 2010, 76, 4858–4862. [Google Scholar] [CrossRef] [Green Version]
- Tanes, C.; Bittinger, K.; Gao, Y.; Friedman, E.S.; Nessel, L.; Paladhi, U.R.; Chau, L.; Panfen, E.; Fischbach, M.A.; Braun, J.; et al. Role of dietary fiber in the recovery of the human gut microbiome and its metabolome. Cell Host Microbe 2021, 29, 394–407.e5. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Yu, X.; Liu, Y.; Sun, L.; Chen, P.; Ding, Q.; Gao, Y.; Zhang, X.; Yu, M.; Liu, Y.; et al. The Baseline Gut Microbiota Directs Dieting-Induced Weight Loss Trajectories. Gastroenterology 2021, 160, 2029–2042.e16. [Google Scholar] [CrossRef] [PubMed]
- Grigor’eva, I.N. Gallstone Disease, Obesity and the Firmicutes/Bacteroidetes Ratio as a Possible Biomarker of Gut Dysbiosis. J. Pers. Med. 2021, 11, 13. [Google Scholar]
- Ferrazzano, G.; Papa, C.; Pollio, A.; Ingenito, A.; Sangianantoni, G.; Cantile, T. Cyanobacteria and Microalgae as Sources of Functional Foods to Improve Human General and Oral Health. Molecules 2020, 25, 5164. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Doud, D.F.R.; Bowers, R.M.; Schulz, F.; De Raad, M.; Deng, K.; Tarver, A.; Glasgow, E.; Meulen, K.V.; Fox, B.; Deutsch, S.; et al. Function-driven single-cell genomics uncovers cellulose-degrading bacteria from the rare biosphere. ISME J. 2019, 14, 659–675. [Google Scholar] [CrossRef] [Green Version]
- Antushevich, H. Fecal microbiota transplantation in disease therapy. Clin. Chim. Acta 2020, 503, 90–98. [Google Scholar] [CrossRef]
- Kim, H.S.; Whon, T.W.; Sung, H.; Jeong, Y.-S.; Jung, E.S.; Shin, N.-R.; Hyun, D.-W.; Kim, P.S.; Lee, J.-Y.; Lee, C.H.; et al. Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 2021, 12, 161. [Google Scholar] [CrossRef]
Figure 1.
Distribution of 16S OTU number of each sample.

Figure 2.
Box-plot representation of alpha diversity. Fecal microbiota were evaluated by the number of observed OTUs (A) and Shannon index (B) between group F and group G.
Figure 2.
Box-plot representation of alpha diversity. Fecal microbiota were evaluated by the number of observed OTUs (A) and Shannon index (B) between group F and group G.
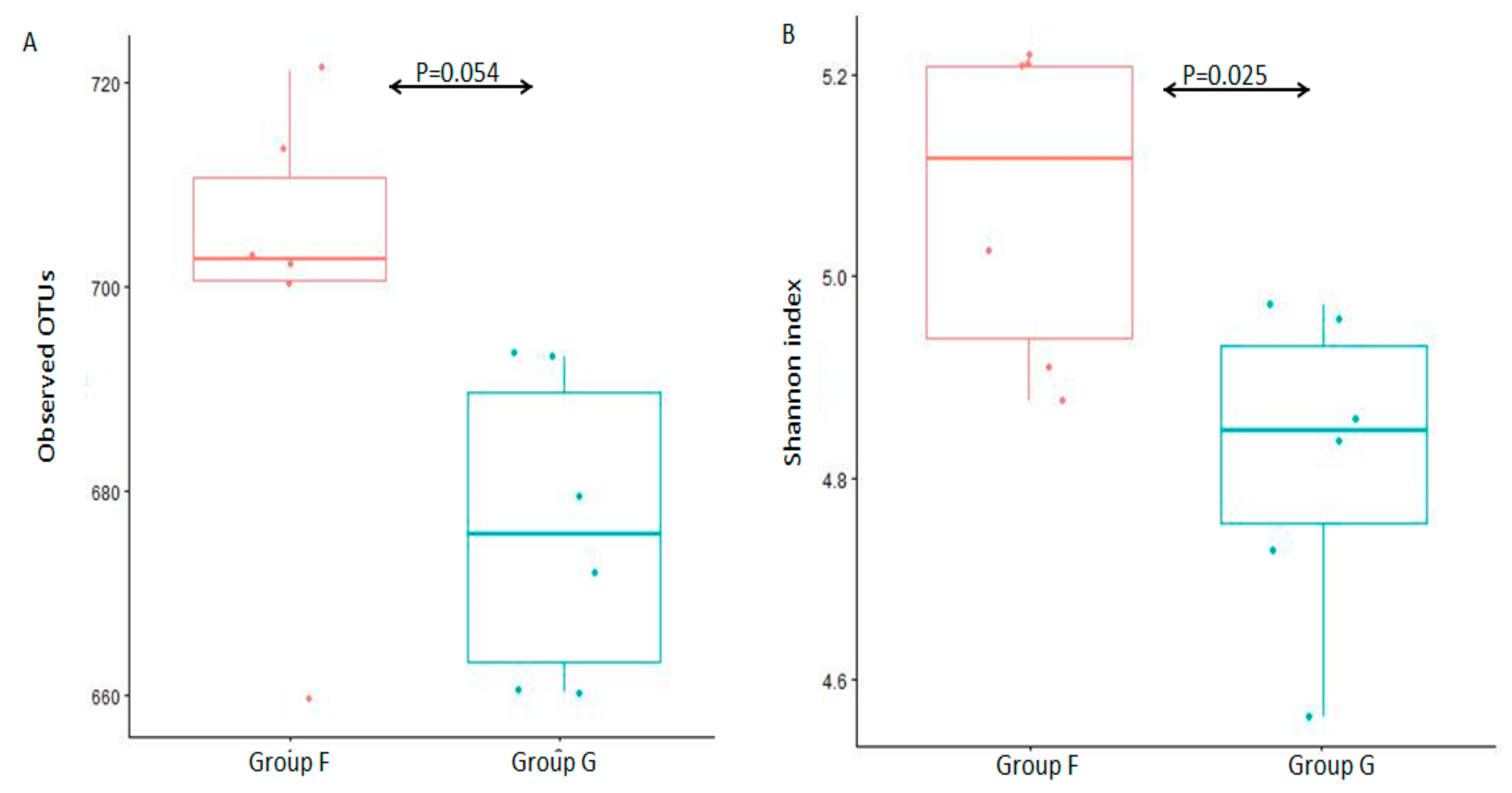
Figure 3.
Principal Coordinates Analysis (PCoA) using Bray-Curtis distance (A) and Jaccard distance (B).
Figure 3.
Principal Coordinates Analysis (PCoA) using Bray-Curtis distance (A) and Jaccard distance (B).

Figure 4.
Relative abundance of fecal microbiota at the phylum level (A) and at the genus level (B).
Figure 4.
Relative abundance of fecal microbiota at the phylum level (A) and at the genus level (B).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, Z.; Yang, L.; He, Y.; Luo, X.; Zhao, S.; Jia, X. Composition of Fecal Microbiota in Grazing and Feedlot Angus Beef Cattle. Animals 2021, 11, 3167. https://doi.org/10.3390/ani11113167
AMA Style
Zhang Z, Yang L, He Y, Luo X, Zhao S, Jia X. Composition of Fecal Microbiota in Grazing and Feedlot Angus Beef Cattle. Animals. 2021; 11(11):3167. https://doi.org/10.3390/ani11113167
Chicago/Turabian StyleZhang, Zhimin, Li Yang, Yang He, Xinmao Luo, Shaokang Zhao, and Xianbo Jia. 2021. "Composition of Fecal Microbiota in Grazing and Feedlot Angus Beef Cattle" Animals 11, no. 11: 3167. https://doi.org/10.3390/ani11113167
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.