
Complete Genome Sequencing of Field Isolates of Peste des Petits Ruminants Virus from Tanzania Revealed a High Nucleotide Identity with Lineage III PPR Viruses

 , , ,
, , ,  , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
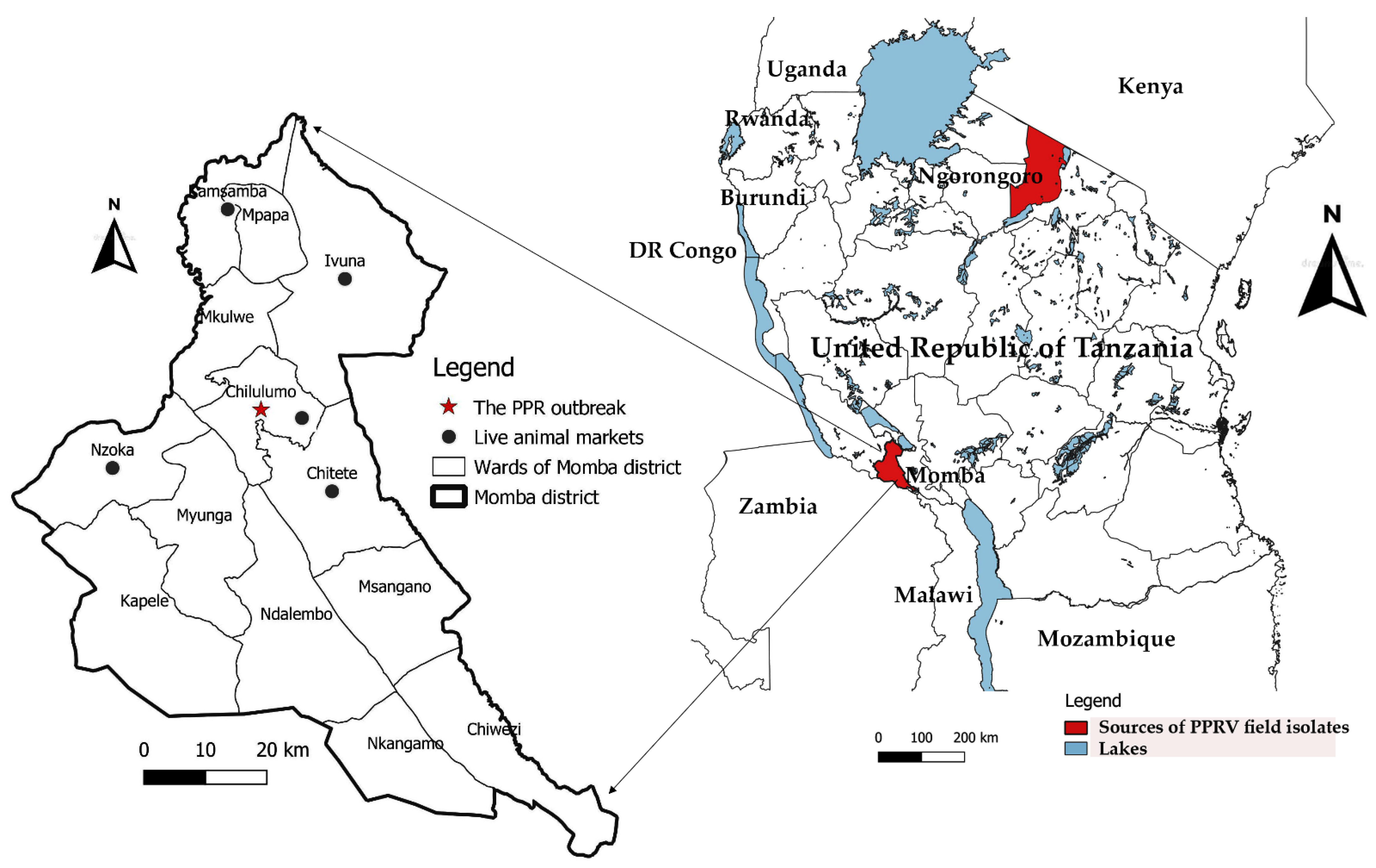
2.1. Sample Source Description and Storage
2.2. Sample Selection
2.3. RNA Extraction, cDNA Synthesis and PCR Amplification
2.4. Nanopore Library Preparation and Sequencing
2.5. Nanopore Dataset Analysis
2.6. Temporal Phylogenetics
3. Results
3.1. Nucleotide Amplification
3.2. Long-Read Nanopore Sequencing of PPRV Field Isolates
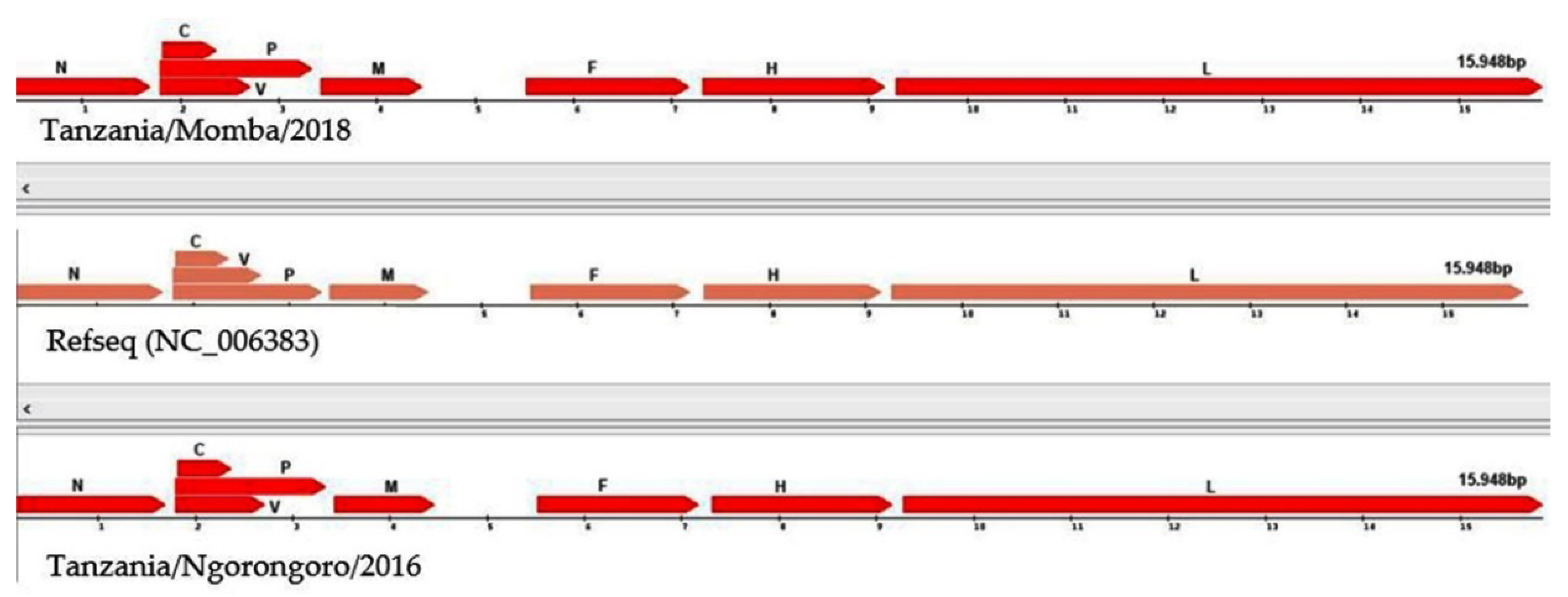
3.3. Annotation of Peste des Petits Ruminants Virus Isolate
3.4. Phylogenetic Analysis of Peste des Petits Ruminants Virus
3.5. Temporal–Spatial Spread of Peste des Petits Ruminants Virus
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Njeumi, F.; Bailey, D.; Soula, J.J.; Diop, B.; Tekola, B.G. Eradicating the Scourge of Peste Des Petits Ruminants from the World. Viruses 2020, 12, 313. [Google Scholar] [CrossRef] [Green Version]
- Libeau, G.; Diallo, A.; Parida, S. Evolutionary Genetics Underlying the Spread of Peste Des Petits Ruminants Virus. Anim. Front. 2014, 4, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Fine, A.E.; Pruvot, M.; Benfield, C.T.O.; Caron, A.; Cattoli, G.; Chardonnet, P.; Dioli, M.; Dulu, T.; Gilbert, M.; Kock, R.; et al. Eradication of Peste Des Petits Ruminants Virus and the Wildlife-Livestock Interface. Front. Vet. Sci. 2020, 7, 50. [Google Scholar] [CrossRef]
- Jones, B.A.; Rich, K.M.; Mariner, J.C.; Anderson, J.; Jeggo, M.; Thevasagayam, S.; Cai, Y.; Peters, A.R.; Roeder, P. The Economic Impact of Eradicating Peste Des Petits Ruminants: A Benefit-Cost Analysis. PLoS ONE 2016, 11, e0149982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO; OIE. Global Strategy for the Control and Eradication of PPR. In Proceedings of the International Conference for the Control and Eradication of Peste Des Petits Ruminants (PPR), Abidjan, Côte d’Ivoire, 31 March–2 April 2015; ISBN 978-92-9044-989-8. [Google Scholar]
- Elsawalhy, A.; Mariner, J.; Chibeu, D.; Wamwayi, H.; Wakhusama, S.; Olaho-Mukani, W.; Toye, P. Pan African Strategy for the Progressive Control of Peste Des Petits Ruminants (Pan African PPR Strategy). Bull. Anim. Health Prod. Afr. 2011. [Google Scholar] [CrossRef]
- Mariner, J.C.; Jones, B.A.; Rich, K.M.; Thevasagayam, S.; Anderson, J.; Jeggo, M.; Cai, Y.; Peters, A.R.; Roeder, P.L. The Opportunity To Eradicate Peste Des Petits Ruminants. J. Immunol. 2016, 196, 3499–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeder, P.; Mariner, J.; Kock, R. Rinderpest: The Veterinary Perspective on Eradication. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120139. [Google Scholar] [CrossRef]
- Parida, S.; Muniraju, M.; Mahapatra, M.; Muthuchelvan, D.; Buczkowski, H.; Banyard, A.C. Peste Des Petits Ruminants. Vet. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dundon, W.G.; Diallo, A.; Cattoli, G. Peste Des Petits Ruminants in Africa: A Review of Currently Available Molecular Epidemiological Data, 2020. Arch. Virol. 2020, 162, 2147–2163. [Google Scholar] [CrossRef]
- Bataille, A.; Kwiatek, O.; Belfkhi, S.; Mounier, L.; Parida, S.; Mahapatra, M.; Caron, A.; Chubwa, C.C.; Keyyu, J.; Kock, R.; et al. Optimization and Evaluation of a Non-Invasive Tool for Peste Des Petits Ruminants Surveillance and Control. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-Time, Portable Genome Sequencing for Ebola Surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Fu, A.; Hu, B.; Tong, Y.; Liu, R.; Liu, Z.; Gu, J.; Xiang, B.; Liu, J.; Jiang, W.; et al. Nanopore Targeted Sequencing for the Accurate and Comprehensive Detection of SARS-CoV-2 and Other Respiratory Viruses. Small 2020, 16, 2002169. [Google Scholar] [CrossRef]
- Faria, N.R.; Quick, J.; Claro, I.M.; Thézé, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; da Costa, A.C.; et al. Establishment and Cryptic Transmission of Zika Virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef]
- Ben Chehida, S.; Filloux, D.; Fernandez, E.; Moubset, O.; Hoareau, M.; Julian, C.; Blondin, L.; Lett, J.-M.; Roumagnac, P.; Lefeuvre, P. Nanopore Sequencing Is a Credible Alternative to Recover Complete Genomes of Geminiviruses. Microorganisms 2021, 9, 903. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and Challenges in Long-Read Sequencing Data Analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Torsson, E.; Kgotlele, T.; Misinzo, G.; Johansson Wensman, J.; Berg, M.; Karlsson Lindsjö, O. Field-Adapted Full Genome Sequencing of Peste-Des-Petits-Ruminants Virus Using Nanopore Sequencing. Front. Vet. Sci. 2020, 7, 858. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, E.P.; Taylor, W.P.; Lawman, M.J.; Bryant, J.; Gibbs, D.P.J.; Taylormichael, W.P.; Lawman, J.P.; Bryant, J. Classification of Peste Des Petits Ruminants Virus as the Fourth Member of the Genus Morbillivirus. Intervirology 1979, 11, 268–274. [Google Scholar] [CrossRef]
- Bailey, D.; Banyard, A.; Dash, P.; Ozkul, A.; Barrett, T. Full Genome Sequence of Peste Des Petits Ruminants Virus, a Member of the Morbillivirus Genus. Virus Res. 2005, 110, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wang, Q.; Zhang, Y.; Liu, C.; Li, L.; Wang, Z. Complete Genome Sequence of a Novel Variant Strain of Peste Des Petits Ruminants Virus, China/XJYL/2013. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, D.; Chard, L.S.; Dash, P.; Barrett, T.; Banyard, A.C. Reverse Genetics for Peste-Des-Petits-Ruminants Virus (PPRV): Promoter and Protein Specificities. Virus Res. 2007, 126, 250–255. [Google Scholar] [CrossRef]
- Mahapatra, M.; Parida, S.; Egziabher, B.G.; Diallo, A.; Barrett, T. Sequence Analysis of the Phosphoprotein Gene of Peste Des Petits Ruminants (PPR) Virus: Editing of the Gene Transcript. Virus Res. 2003, 96, 85–98. [Google Scholar] [CrossRef]
- Balamurugan, V.; Roy, M.; Sowjanyakumari, S.; Abraham, S.; Apsana, R.; Nagalingam, M.; Hemadri, D.; Rahman, H. Development of Recombinant Nucleocapsid Protein Based Indirect ELISA for Serodiagnosis of Peste Des Petits Ruminants in Sheep and Goats. Adv. Anim. Vet. Sci. 2016, 4, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Kwiatek, O.; Minet, C.; Grillet, C.; Hurard, C.; Carlsson, E.; Karimov, B.; Albina, E.; Diallo, A.; Libeau, G. Peste Des Petits Ruminants (PPR) Outbreak in Tajikistan. J. Comp. Pathol. 2007, 136, 111–119. [Google Scholar] [CrossRef]
- Kwiatek, O.; Ali, Y.H.; Saeed, I.K.; Khalafalla, A.I.; Mohamed, O.I.; Obeida, A.A.; Abdelrahman, M.B.; Osman, H.M.; Taha, K.M.; Abbas, Z.; et al. Asian Lineage of Peste Des Petits Ruminants Virus, Africa. Emerg. Infect. Dis. 2011, 17, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Couacy-Hymann, E.; Roger, F.; Hurard, C.; Guillou, J.P.; Libeau, G.; Diallo, A. Rapid and Sensitive Detection of Peste Des Petits Ruminants Virus by a Polymerase Chain Reaction Assay. J. Virol. Methods 2002, 100, 17–25. [Google Scholar] [CrossRef]
- Forsyth, M.A.; Barrett, T. Evaluation of Polymerase Chain Reaction for the Detection and Characterisation of Rinderpest and Peste Des Petits Ruminants Viruses for Epidemiological Studies. Virus Res. 1995, 39, 151–163. [Google Scholar] [CrossRef]
- Muniraju, M.; Harrak, M.E.; Bao, J.; Parthiban, A.B.R.; Banyard, A.C.; Batten, C.; Parida, S. Complete Genome Sequence of a Peste Des Petits Ruminants Virus Recovered from an Alpine Goat during an Outbreak in Morocco in 2008. Genome Announc. 2013, 1, e00096-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baazizi, R.; Mahapatra, M.; Clarke, B.D.; Ait-Oudhia, K.; Khelef, D.; Parida, S. Peste Des Petits Ruminants (PPR): A Neglected Tropical Disease in Maghreb Region of North Africa and Its Threat to Europe. PLoS ONE 2017, 12, e0175461. [Google Scholar] [CrossRef] [PubMed]
- Maganga, G.D.; Verrier, D.; Zerbinati, R.M.; Drosten, C.; Drexler, J.F.; Leroy, E.M. Molecular Typing of PPRV Strains Detected during an Outbreak in Sheep and Goats in South-Eastern Gabon in 2011. Virol. J. 2013, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.S.; Babu, A.; Sundarapandian, G.; Roy, P.; Thangavelu, A.; Kumar, K.S.; Arumugam, R.; Banyard, A.C.; Manohar, B.M.; Parida, S. Molecular Characterisation of Lineage IV Peste Des Petits Ruminants Virus Using Multi Gene Sequence Data. Vet. Microbiol. 2014, 174, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Woma, T.Y.; Qasim, A.M.M.; Sabi, A.A.; Abraham, M.N.; Olaiya, O.D.; Bailey, D.; Shamaki, D. Co-Circulation of Peste-Des-Petits-Ruminants Virus Asian Lineage IV with Lineage II in Nigeria. Transbound. Emerg. Dis. 2016, 63, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Wambura, P. Serological Evidence of the Absence of Peste Des Petits Ruminants in Tanzania. Vet. Rec. 2000, 146, 473–474. [Google Scholar] [CrossRef]
- Swai, E.S.; Kapaga, A.; Kivaria, F.; Tinuga, D.; Joshua, G.; Sanka, P. Prevalence and Distribution of Peste Des Petits Ruminants Virus Antibodies in Various Districts of Tanzania. Vet. Res. Commun. 2009, 33, 927–936. [Google Scholar] [CrossRef]
- Karimuribo, E.D.; Loomu, P.M.; Mellau, L.S.B.; Swai, E.S. Retrospective Study on Sero-Epidemiology of Peste Des Petits Ruminants before Its Official Confirmation in Northern Tanzania in 2008. Res. Opin. Anim. Vet. Sci. 2010, 1, 184–187. [Google Scholar]
- Kinimi, E.; Odongo, S.; Muyldermans, S.; Kock, R.; Misinzo, G. Paradigm Shift in the Diagnosis of Peste Des Petits Ruminants: Scoping Review. Acta Vet. Scand. 2020, 62, 7. [Google Scholar] [CrossRef]
- Diallo, A.; Barrett, T.; Barbron, M.; Meyer, G.; Lefevre, P.C. Cloning of the Nucleocapsid Protein Gene of Peste-Des-Petits-Ruminants Virus: Relationship to Other Morbilliviruses. J. Gen. Virol. 1994, 75, 233–237. [Google Scholar] [CrossRef]
- Meyer, G.; Diallo, A. The Nucleotide Sequence of the Fusion Protein Gene of the Peste Des Petits Ruminants Virus: The Long Untranslated Region in the 5′-End of the F-Protein Gene of Morbilliviruses Seems to Be Specific to Each Virus. Virus Res. 1995, 37, 23–35. [Google Scholar] [CrossRef]
- Kwiatek, O.; Keita, D.; Gil, P.; Fernández-Pinero, J.; Jimenez Clavero, M.A.; Albina, E.; Libeau, G. Quantitative One-Step Real-Time RT-PCR for the Fast Detection of the Four Genotypes of PPRV. J. Virol. Methods 2010, 165, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Kivaria, F.M.; Kwiatek, O.; Kapaga, A.M.; Swai, E.S.; Libeau, G.; Moshy, W.; Mbyuzi, A.O.; Gladson, J. The Incursion, Persistence and Spread of Peste Des Petits Ruminants in Tanzania: Epidemiological Patterns and Predictions. Onderstepoort J. Vet. Res. 2014, 80, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muse, E.A.; Karimuribo, E.D.; Gitao, G.C.; Misinzo, G.; Mellau, L.S.B.; Msoffe, P.L.M.; Swai, E.S.; Albano, M.O. Epidemiological Investigation into the Introduction and Factors for Spread of Peste Des Petits Ruminants, Southern Tanzania. Onderstepoort J. Vet. Res. 2012, 79, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kgotlele, T.; Kasanga, C.J.; Kusiluka, L.J.M.; Misinzo, G. Preliminary Investigation on Presence of Peste Des Petits Ruminants in Dakawa, Mvomero District, Morogoro Region, Tanzania. Onderstepoort J. Vet. Res. 2014, 81, 1–3. [Google Scholar] [CrossRef]
- Kgotlele, T.; Macha, E.S.; Kasanga, C.J.; Kusiluka, L.J.M.; Karimuribo, E.D.; Van Doorsselaere, J.; Wensman, J.J.; Munir, M.; Misinzo, G. Partial Genetic Characterization of Peste Des Petits Ruminants Virus from Goats in Northern and Eastern Tanzania. Transbound. Emerg. Dis. 2014, 61, 56–62. [Google Scholar] [CrossRef]
- Mahapatra, M.; Sayalel, K.; Muniraju, M.; Eblate, E.; Fyumagwa, R.; Shilinde, L.; Mdaki, M.; Keyyu, J.; Parida, S.; Kock, R. Spillover of Peste Des Petits Ruminants Virus from Domestic to Wild Ruminants in the Serengeti Ecosystem, Tanzania. Emerg. Infect. Dis. 2015, 21, 2230–2234. [Google Scholar] [CrossRef] [Green Version]
- Misinzo, G.; Kgotlele, T.; Muse, E.; Doorsselaere, J.; Berg, M.; Munir, M. Peste Des Petits Ruminants Virus Lineage II and IV From Goats in Southern Tanzania During an Outbreak in 2011. Br. J. Virol. 2015, 2, 1–4. [Google Scholar]
- Jones, B.A.; Mahapatra, M.; Chubwa, C.; Clarke, B.; Batten, C.; Hicks, H.; Henstock, M.; Keyyu, J.; Kock, R.; Parida, S. Characterisation of Peste Des Petits Ruminants Disease in Pastoralist Flocks in Ngorongoro District of Northern Tanzania and Bluetongue Virus Co-Infection. Viruses 2020, 12, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kgotlele, T.; Chota, A.; Chubwa, C.C.; Nyasebwa, O.; Lyimo, B.; Torsson, E.; Karimuribo, E.; Kasanga, C.J.; Wensman, J.J.; Misinzo, G.; et al. Detection of Peste Des Petits Ruminants and Concurrent Secondary Diseases in Sheep and Goats in Ngorongoro District, Tanzania. Comp. Clin. Pathol. 2019, 28, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Chota, A.; Shirima, G.; Kusiluka, L. Detection of Contagious Caprine Pleuropneumonia and Concurrent Diseases in Outbreaks Presenting with Respiratory Signs in Small Ruminants in Tanzania. Int. J. Trop. Dis. Health 2020. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2016 Update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing Bacterial Genome Assemblies with Multiplex MinION Sequencing. Microb. Genomics 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and Processing Long-Read Sequencing Data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and Accurate Long-Read Assembly via Adaptive κ-Mer Weighting and Repeat Separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and Accurate de Novo Genome Assembly from Long Uncorrected Reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Genome Annotation Transfer Utility (GATU): Rapid Annotation of Viral Genomes Using a Closely Related Reference Genome | BMC Genomics | Full Text. Available online: https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-7-150 (accessed on 8 October 2021).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Muniraju, M.; Munir, M.; Parthiban, A.R.; Banyard, A.C.; Bao, J.; Wang, Z.; Ayebazibwe, C.; Ayelet, G.; El Harrak, M.; Mahapatra, M.; et al. Molecular Evolution of Peste Des Petits Ruminants Virus. Emerg. Infect. Dis. 2014. [Google Scholar] [CrossRef] [Green Version]
- Dundon, W.G.; Kihu, S.M.; Settypalli, T.B.K.; Gitao, G.C.; Bebora, L.C.; John, N.M.; Oyugi, J.O.; Silber, R.; Loitsch, A.; Diallo, A. First Complete Genome Sequence of a Lineage III Peste Des Petits Ruminants Virus. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Niyokwishimira, A.; de D Baziki, J.; Dundon, W.G.; Nwankpa, N.; Njoroge, C.; Boussini, H.; Wamwayi, H.; Jaw, B.; Cattoli, G.; Nkundwanayo, C.; et al. Detection and Molecular Characterization of Peste Des Petits Ruminants Virus from Outbreaks in Burundi, December 2017–January 2018. Transbound. Emerg. Dis. 2019, 66, 2067–2073. [Google Scholar] [CrossRef] [PubMed]
- Muniraju, M.; Munir, M.; Banyard, A.C.; Ayebazibwe, C.; Wensman, J.; Zohari, S.; Berg, M.; Parthiban, A.B.R.; Mahapatra, M.; Libeau, G.; et al. Complete Genome Sequences of Lineage III Peste Des Petits Ruminants Viruses from the Middle East and East Africa. Genome Announc. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Chen, W.; Huang, K.; Baron, M.D.; Bu, Z. Rescue of Recombinant Peste Des Petits Ruminants Virus: Creation of a GFP-Expressing Virus and Application in Rapid Virus Neutralization Test. Vet. Res. 2012, 43, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Adombi, C.M.; Waqas, A.; Dundon, W.G.; Li, S.; Daojin, Y.; Kakpo, L.; Aplogan, G.L.; Diop, M.; Lo, M.M.; Silber, R.; et al. Peste Des Petits Ruminants in Benin: Persistence of a Single Virus Genotype in the Country for Over 42 Years. Transbound. Emerg. Dis. 2017, 64, 1037–1044. [Google Scholar] [CrossRef]
- Chard, L.S.; Bailey, D.S.; Dash, P.; Banyard, A.C.; Barrett, T. Full Genome Sequences of Two Virulent Strains of Peste-Des-Petits Ruminants Virus, the Côte d’Ivoire 1989 and Nigeria 1976 Strains. Virus Res. 2008, 136, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Dundon, W.G.; Adombi, C.; Waqas, A.; Otsyina, H.R.; Arthur, C.T.; Silber, R.; Loitsch, A.; Diallo, A. Full Genome Sequence of a Peste Des Petits Ruminants Virus (PPRV) from Ghana. Virus Genes 2014, 49, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Muniraju, M.; Mahapatra, M.; Ayelet, G.; Babu, A.; Olivier, G.; Munir, M.; Libeau, G.; Batten, C.; Banyard, A.C.; Parida, S. Emergence of Lineage IV Peste Des Petits Ruminants Virus in Ethiopia: Complete Genome Sequence of an Ethiopian Isolate 2010. Transbound. Emerg. Dis. 2016, 63, 435–442. [Google Scholar] [CrossRef]
- Rajko-Nenow, P.Z.; Cunliffe, T.G.; Flannery, J.T.; Ropiak, H.M.; Avaliani, L.; Donduashvili, M.; Baron, M.D.; Batten, C.A. Complete Genome Sequence of Peste Des Petits Ruminants Virus from Georgia, 2016. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacıoğlu, S.; King, S.; Çizmeci, Ş.G.; Yeşil, Ö.; Flannery, J.; Baron, M.D.; Batten, C.; Rajko-Nenow, P.Z. Complete Genome Sequence of a Lineage IV Peste Des Petits Ruminants Virus from Turkey, 2018. Microbiol. Resour. Announc. 2020. [Google Scholar] [CrossRef] [Green Version]
- Shatar, M.; Khanui, B.; Purevtseren, D.; Khishgee, B.; Loitsch, A.; Unger, H.; Settypalli, T.B.K.; Cattoli, G.; Damdinjav, B.; Dundon, W.G. First Genetic Characterization of Peste Des Petits Ruminants Virus from Mongolia. Arch. Virol. 2017, 162, 3157–3160. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Wang, Q.; Li, L.; Liu, C.; Zhang, Z.; Li, J.; Wang, S.; Wu, X.; Wang, Z. Evolutionary Dynamics of Recent Peste Des Petits Ruminants Virus Epidemic in China during 2013–2014. Virology 2017, 510, 156–164. [Google Scholar] [CrossRef]
- Li, L.; Bao, J.; Wu, X.; Wang, Z.; Wang, J.; Gong, M.; Liu, C.; Li, J. Rapid Detection of Peste Des Petits Ruminants Virus by a Reverse Transcription Loop-Mediated Isothermal Amplification Assay. J. Virol. Methods 2010, 170, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Munir, M.; Shabbir, M.Z. A Comparative Phylogenomic Analysis of Peste Des Petits Ruminants Virus Isolated from Wild and Unusual Hosts. Mol. Biol. Rep. 2019, 46, 5587–5593. [Google Scholar] [CrossRef]
- Eloiflin, R.J.; Boyer, M.; Kwiatek, O.; Guendouz, S.; Loire, E.; Servan de Almeida, R.; Libeau, G.; Bataille, A. Evolution of Attenuation and Risk of Reversal in Peste Des Petits Ruminants Vaccine Strain Nigeria 75/1. Viruses 2019, 11, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC Multiplex PCR Method for SARS-CoV-2 Genome Sequencing Using Nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mamedov, T.G.; Pienaar, E.; Whitney, S.E.; TerMaat, J.R.; Carvill, G.; Goliath, R.; Subramanian, A.; Viljoen, H.J. A Fundamental Study of the PCR Amplification of GC-Rich DNA Templates. Comput. Biol. Chem. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, M.; El-sayed, A. Toward Peste Des Petits Virus ( PPRV ) Eradication: Diagnostic Approaches, Novel Vaccines, and Control Strategies. Virus Res. 2019, 274, 197774. [Google Scholar] [CrossRef] [PubMed]
- Benfield, C.T.O.; Hill, S.; Shatar, M.; Shiilegdamba, E.; Damdinjav, B.; Fine, A.; Willett, B.; Kock, R.; Bataille, A. Molecular Epidemiology of Peste Des Petits Ruminants Virus Emergence in Critically Endangered Mongolian Saiga Antelope and Other Wild Ungulates. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kinimi, E.; Muyldermans, S.; Vincke, C.; Odongo, S.; Kock, R.; Parida, S.; Mahapatra, M.; Misinzo, G. Development of Nanobodies Targeting Peste Des Petits Ruminants Virus: The Prospect in Disease Diagnosis and Therapy. Animals 2021, 11, 2206. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.A.; Mahapatra, M.; Mdetele, D.; Keyyu, J.; Gakuya, F.; Eblate, E.; Lekolool, I.; Limo, C.; Ndiwa, J.N.; Hongo, P.; et al. Peste Des Petits Ruminants Virus Infection at the Wildlife–Livestock Interface in the Greater Serengeti Ecosystem, 2015–2019. Viruses 2021, 13, 838. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region/District | Study Period | Host | Sequence | Lineage | References |
|---|---|---|---|---|---|
| Arusha, Kilimanjaro, Manyara and Tanga | 2010 | sheep and goats | partial | III | [40] |
| Tandahimba and Newala | 2011 | sheep and goats | - | - | [41] |
| Mvomero | 2013 | sheep and goats | - | - | [42] |
| Ngorongoro and Mvomero | 2013 | goats | partial | III | [43] |
| Ngorongoro | 2014 | sheep and Grant’s gazelle | partial | II | [44] |
| Tandahimba | 2015 | sheep and goats | partial | II and IV | [45] |
| Ngorongoro | 2015 | sheep and goats | partial | III | [46] |
| Ngorongoro | 2016 | sheep and goats | - | - | [47] |
| Mbeya, Iringa, Dodoma Morogoro, Pwani Serengeti, Tanga and Arusha | 2018 | sheep and goats | - | - | [48] |
| Mvomero | 2020 | sheep and goats | complete | III | [17] |
| Primer Name | Fragments | Sequence (5′ to 3′) | Tm * | GC% | Position on Genome | Fragment Size (bp) | |
|---|---|---|---|---|---|---|---|
| Start | End | ||||||
| PPRV7LK | F7 | CAACAACACTCCGCTGTCCT | 60.30 | 55.00 | 3778 | 3796 | 770 |
| PPRV7RK | GAGTGGCTGTGTTGGTGCT | 60.53 | 57.89 | 4548 | 4530 | ||
| PPRV8LK | F8 | CAAGCCGTCCTACAGCCATC | 60.81 | 60.00 | 4353 | 4372 | 895 |
| PPRV8RK | GTCCTCCCTCGGTCTGTCT | 60.00 | 63.16 | 5248 | 5229 | ||
| PPRV9LK | F9 | GAGGACACCCAACCACCGAAAC | 59.00 | 59.09 | 4972 | 4993 | 815 |
| PPRV9RK | ACAGAGCATCCTCTACAGGCTT | 55.00 | 50.00 | 5787 | 5766 | ||
| Sample | Raw Reads | Total bp | N50 Length (bp) | Reads Mapped to PPRV | Average Coverage Reads | Genome Coverage >50× (%) | Genome Coverage >25× (%) Source | PPRV Lineage |
|---|---|---|---|---|---|---|---|---|
| Ngorongoro | 1,881,426 | 2,203,973,564 | 793 | 1,784,633 | 4575 | 99.2 | 99.8 | III |
| Momba | 1,712,393 | 1,605,543,198 | 816 | 1,688,419 | 3906 | 98.7 | 99.5 | III |
| Isolate Name | GenBank | Country of Origin | Year of Collection | Lineage | Percentage Nucleotide Identity with Tanzania /2016 PPRV Isolate | Percentage Nucleotide Identity with Tanzania/ 2018 PPRV Isolate | Host Species | Reference |
|---|---|---|---|---|---|---|---|---|
| Tanzania/2016 | MW960272 | Tanzania | 2016 | III | 100.00 | 97.39 | goat | This study |
| Tanzania/2018 | MZ322753 | Tanzania | 2018 | III | 97.39 | 100.00 | goat | This study |
| KN5/2011 | KM463083.1 | Kenya | 2011 | III | 99.24 | 97.92 | goat | [60] |
| B3 | MK686066.1 | Burundi | 2017 | III | 98.44 | 97.22 | goat | [61] |
| Uganda 2012 | KJ867543.1 | Uganda | 2012 | III | 97.38 | 96.19 | goat | [62] |
| Ethiopia 1994 | KJ867540.1 | Ethiopia | 1994 | III | 95.55 | 95.57 | goat | [62] |
| UAE 1986 | KJ867545.1 | United Arab Emirates | 1986 | III | 94.49 | 94.57 | Dorcas gazelle | [62] |
| Oman 1983 | KJ867544.1 | Oman | 1983 | III | 94.47 | 94.56 | goat | [62] |
| Nigeria/75/1 | HQ197753.1 | Nigeria | 1976 | II | 88.58 | 88.73 | goat | [63] |
| Benin/B1/1969 | KR781450.1 | Benin | 1969 | II | 88.85 | 89.00 | goat | [64] |
| Ng76/1 | EU267274.1 | Nigeria | 1976 | II | 88.50 | 88.66 | goat | [65] |
| Ghana/2010 | KJ466104.1 | Ghana | 2010 | II | 87.69 | 87.81 | sheep | [66] |
| Benin/10/2011 | KR781449.1 | Benin | 2011 | II | 87.59 | 87.72 | sheep | [64] |
| ICV89 | EU267273.1 | Cote d’Ivoire | 1989 | I | 87.92 | 88.02 | goat | [65] |
| Ethiopia 2010 | KJ867541.1 | Ethiopia | 2010 | IV | 87.20 | 87.35 | goat | [67] |
| Georgia/2016 | MF737202.1 | Georgia | 2016 | IV | 86.94 | 87.04 | sheep | [68] |
| Morocco 2008 | KC594074.1 | Morocco | 2008 | IV | 87.35 | 87.44 | goat | [28] |
| Turkey/2018 | MN657232.1 | Turkey | 2018 | IV | 86.85 | 87.01 | sheep | [69] |
| Mongolia/2016 | KY888168.1 | Mongolia | 2016 | IV | 86.86 | 86.88 | sheep | [70] |
| China/33/2007 | KX421388.1 | China | 2007 | IV | 87.30 | 87.36 | goat | [71] |
| China/Tibet/07 | FJ905304.1 | China | 2007 | IV | 87.28 | 87.34 | goat | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinimi, E.; Mahapatra, M.; Kgotlele, T.; Makange, M.R.; Tennakoon, C.; Njeumi, F.; Odongo, S.; Muyldermans, S.; Kock, R.; Parida, S.; et al. Complete Genome Sequencing of Field Isolates of Peste des Petits Ruminants Virus from Tanzania Revealed a High Nucleotide Identity with Lineage III PPR Viruses. Animals 2021, 11, 2976. https://doi.org/10.3390/ani11102976
Kinimi E, Mahapatra M, Kgotlele T, Makange MR, Tennakoon C, Njeumi F, Odongo S, Muyldermans S, Kock R, Parida S, et al. Complete Genome Sequencing of Field Isolates of Peste des Petits Ruminants Virus from Tanzania Revealed a High Nucleotide Identity with Lineage III PPR Viruses. Animals. 2021; 11(10):2976. https://doi.org/10.3390/ani11102976
Chicago/Turabian StyleKinimi, Edson, Mana Mahapatra, Tebogo Kgotlele, Mariam R. Makange, Chandana Tennakoon, Felix Njeumi, Steven Odongo, Serge Muyldermans, Richard Kock, Satya Parida, and et al. 2021. "Complete Genome Sequencing of Field Isolates of Peste des Petits Ruminants Virus from Tanzania Revealed a High Nucleotide Identity with Lineage III PPR Viruses" Animals 11, no. 10: 2976. https://doi.org/10.3390/ani11102976
APA StyleKinimi, E., Mahapatra, M., Kgotlele, T., Makange, M. R., Tennakoon, C., Njeumi, F., Odongo, S., Muyldermans, S., Kock, R., Parida, S., Rweyemamu, M., & Misinzo, G. (2021). Complete Genome Sequencing of Field Isolates of Peste des Petits Ruminants Virus from Tanzania Revealed a High Nucleotide Identity with Lineage III PPR Viruses. Animals, 11(10), 2976. https://doi.org/10.3390/ani11102976