
Species-Level Gut Microbiota Analysis after Antibiotic-Induced Dysbiosis in Horses


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Animals
2.2. Microbiota Analysis for PacBio and Illumina Sequencing
2.3. Sequence Analysis
2.4. Statistical Analysis
3. Results
3.1. Horses
3.2. Characterization of the Horse Fecal Microbiota Using Long-Read Sequencing
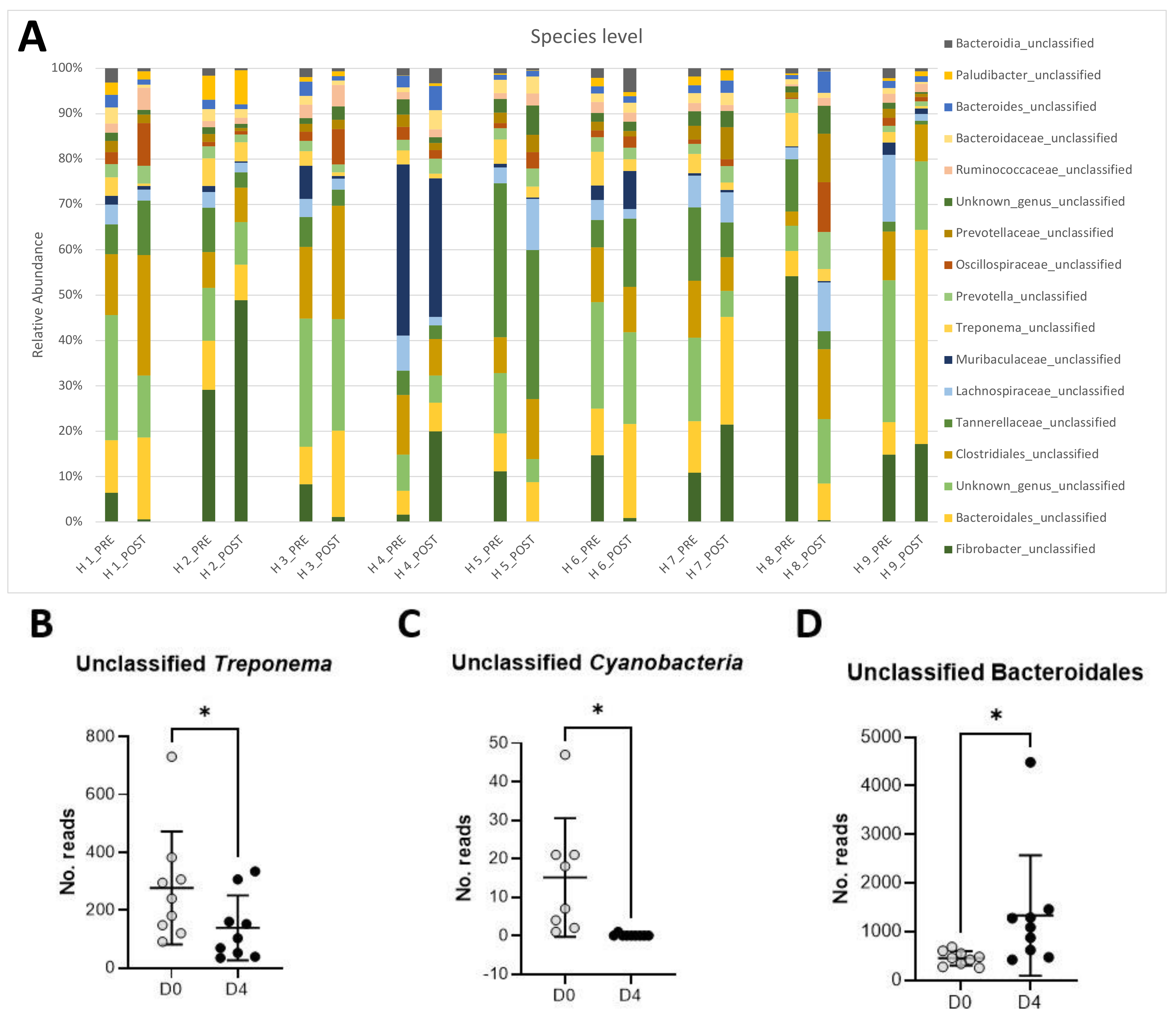
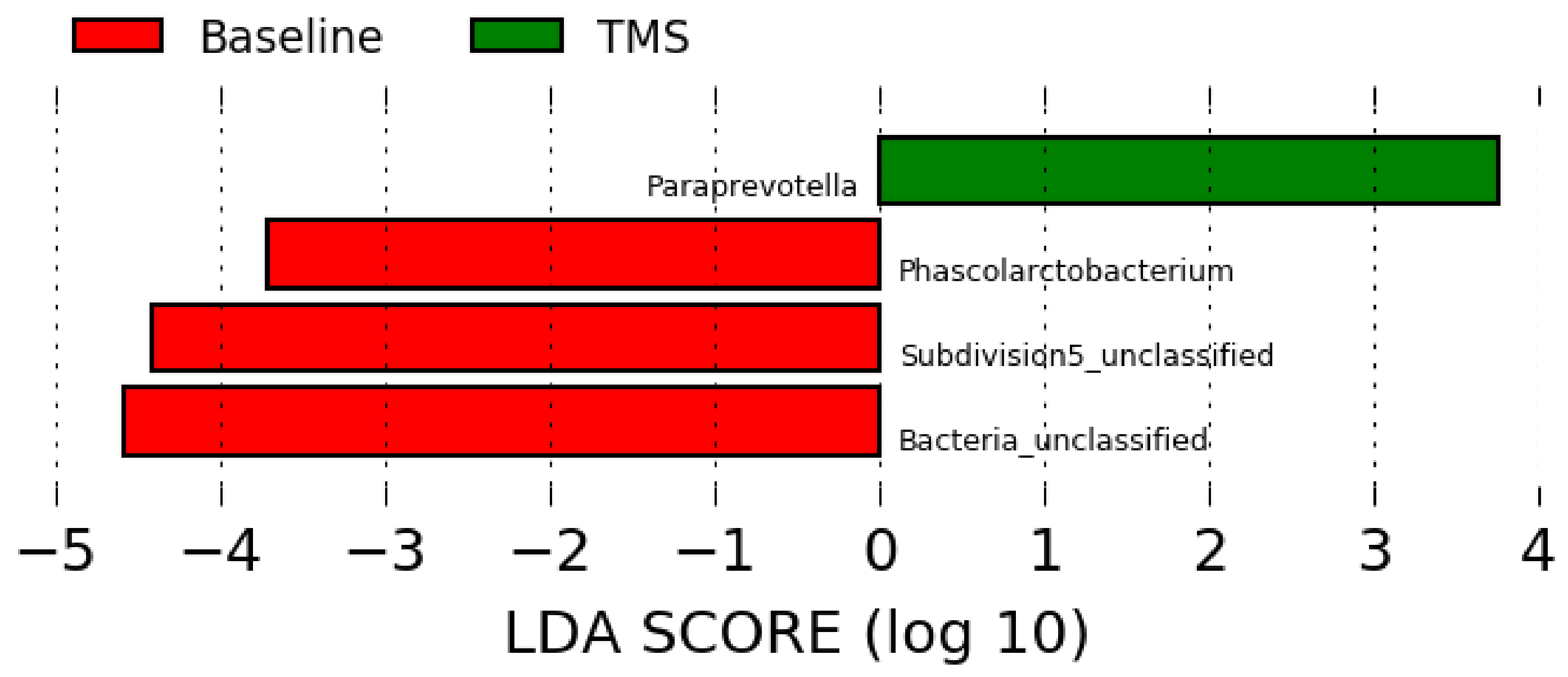
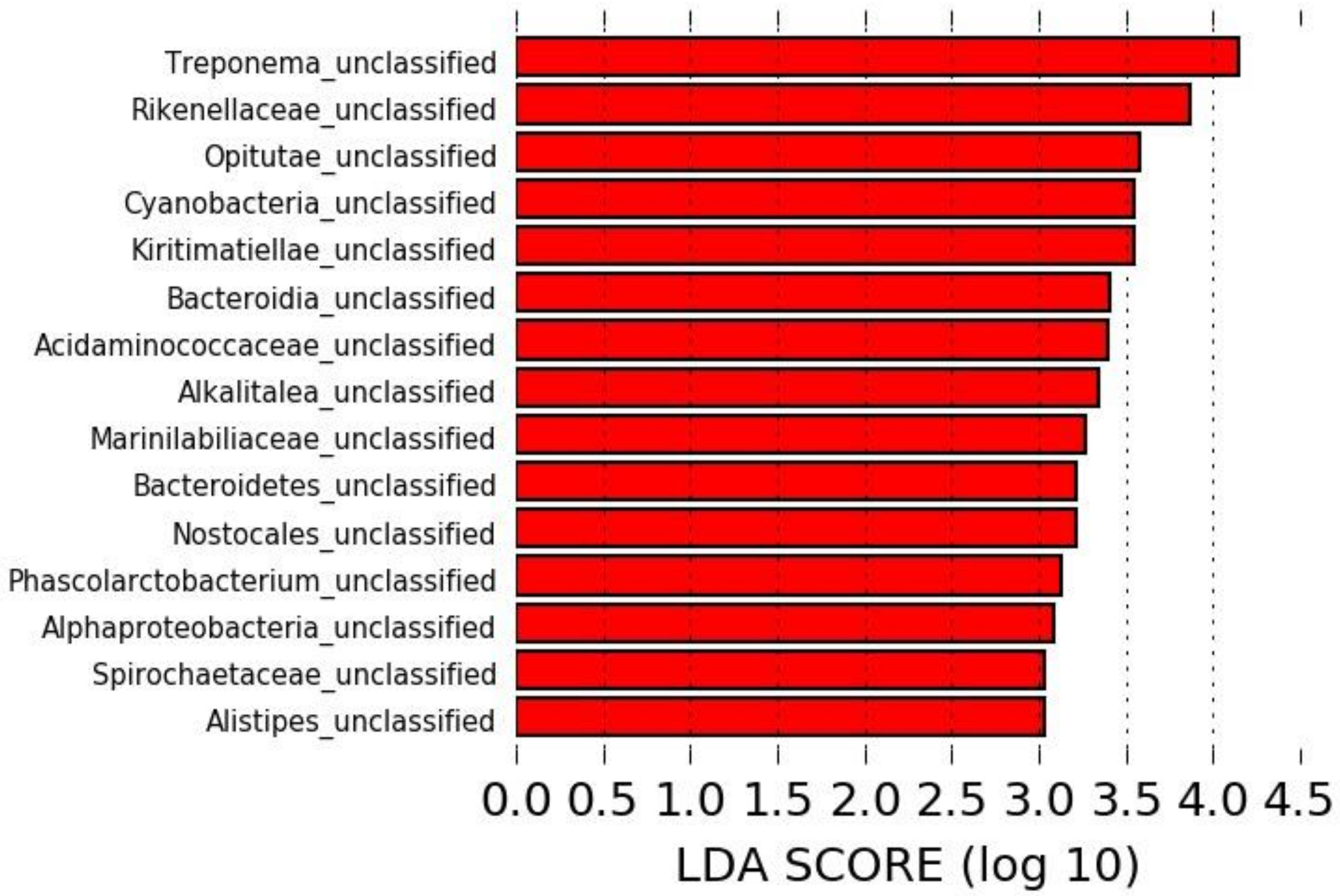
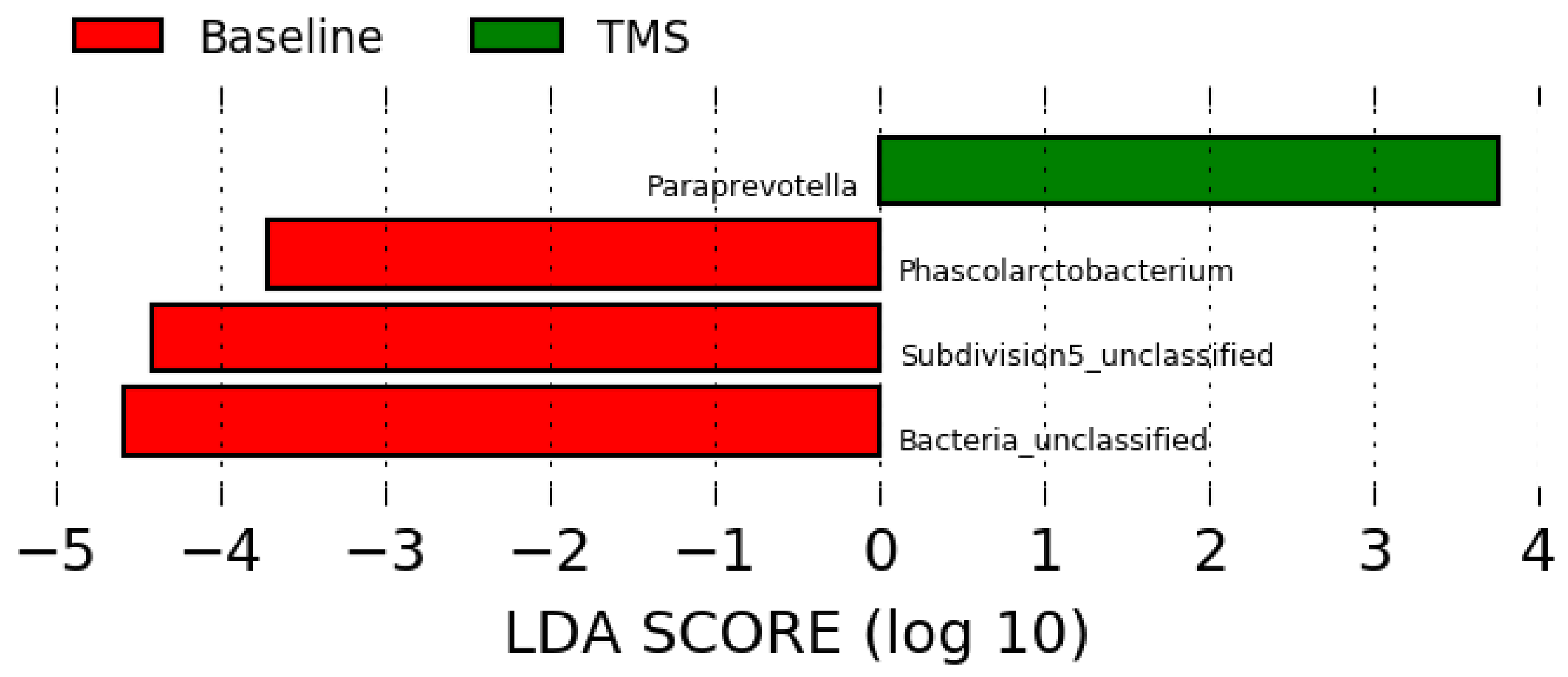
3.3. Long-Read Microbiota Analysis after TMS Administration
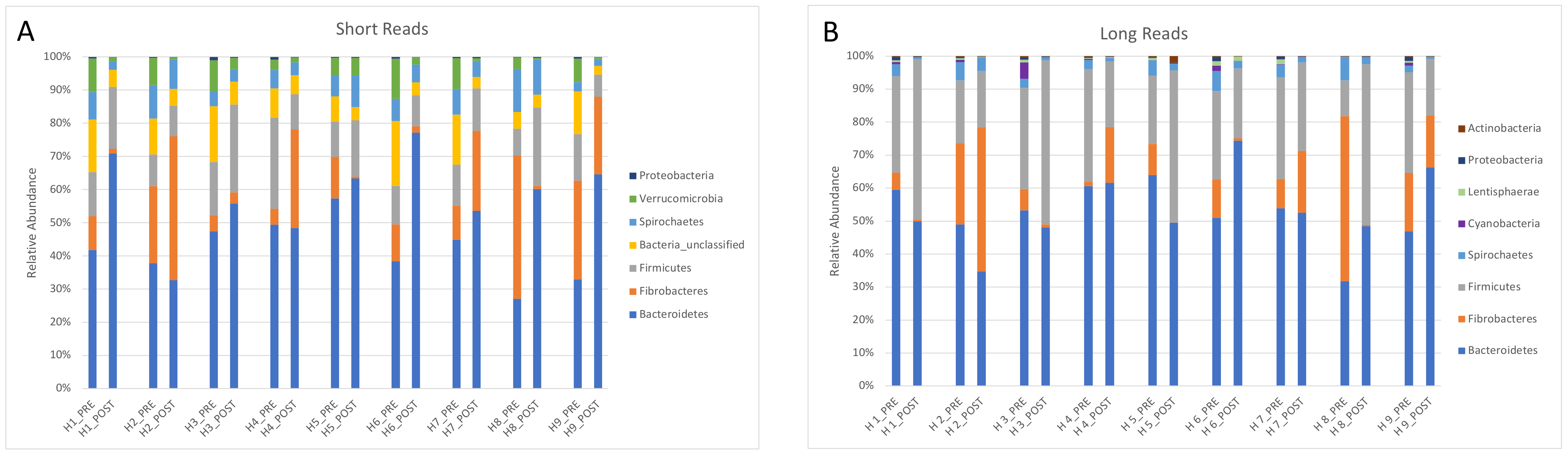
3.4. Comparison between Two Sequencing Protocols (Illumina Short-Read and PacBio Long-Read)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, M.J.; Smith, R.M.; Spires, H.R.; Davis, C.L. Measurement of Volatile Fatty Acid Production Rates in the Cecum of the Pony. J. Anim. Sci. 1976, 42, 1465–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stämpfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, J.S. Comparison of the Fecal Microbiota of Healthy Horses and Horses with Colitis by High Throughput Sequencing of the V3–V5 Region of the 16S rRNA Gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney, C.A.; Bedenice, D.; Pacheco, A.P.; Oliveira, B.C.M.; Paradis, M.-R.; Mazan, M.; Widmer, G. Assessment of clinical and microbiota responses to fecal microbial transplantation in adult horses with diarrhea. PLoS ONE 2021, 16, e0244381. [Google Scholar] [CrossRef]
- Stewart, H.L.; Pitta, D.; Indugu, N.; Vecchiarelli, B.; Hennessy, M.L.; Engiles, J.B.; Southwood, L.L. Changes in the faecal bacterial microbiota during hospitalisation of horses with colic and the effect of different causes of colic. Equine Veter. J. 2020. [Google Scholar] [CrossRef]
- Costa, M.C.; Stämpfli, H.R.; Arroyo, L.G.; Allen-Vercoe, E.; Gomes, R.G.; Weese, J.S. Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs. BMC Veter. Res. 2015, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.; Pilla, R.; Chaffin, K.; Lidbury, J.; Steiner, J.; Suchodolski, J. Alterations in the Fecal Microbiome and Metabolome of Horses with Antimicrobial-Associated Diarrhea Compared to Antibiotic-Treated and Non-Treated Healthy Case Controls. Animals 2021, 11, 1807. [Google Scholar] [CrossRef]
- Volkova, A.; Ruggles, K.; Schulfer, A.; Gao, Z.; Ginsberg, S.D.; Blaser, M.J. Effects of early-life penicillin exposure on the gut microbiome and frontal cortex and amygdala gene expression. iScience 2021, 24, 102797. [Google Scholar] [CrossRef]
- Arnold, C.E.; Isaiah, A.; Pilla, R.; Lidbury, J.; Coverdale, J.S.; Callaway, T.R.; Lawhon, S.D.; Steiner, J.; Suchodolski, J.S. The cecal and fecal microbiomes and metabolomes of horses before and after metronidazole administration. PLoS ONE 2020, 15, e0232905. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J. Growing Unculturable Bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.C.; Weese, J.S. Understanding the Intestinal Microbiome in Health and Disease. Veter. Clin. North Am. Equine Pract. 2018, 34, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Weese, J.S. Methods and basic concepts for microbiota assessment. Veter. J. 2019, 249, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whon, T.W.; Chung, W.-H.; Lim, M.Y.; Song, E.-J.; Kim, P.S.; Hyun, D.-W.; Shin, N.-R.; Bae, J.-W.; Nam, Y.-D. The effects of sequencing platforms on phylogenetic resolution in 16 S rRNA gene profiling of human feces. Sci. Data 2018, 5, 180068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, J.; Ledala, N.; Caimano, M.J.; Jackson, E.; Gratalo, D.; Fasulo, D.; Driscoll, M.D.; Coleman, S.; Matson, A.P. High-Resolution Differentiation of Enteric Bacteria in Premature Infant Fecal Microbiomes Using a Novel rRNA Amplicon. mBio 2021, 12, e03656-20. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2016, 1, e00009-15. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.; Highlander, S.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowsky, M.J.; Staley, C.; Heiner, C.; Hall, R.; Kelly, C.R.; Brandt, L.; Khoruts, A. Analysis of gut microbiota—An ever changing landscape. Gut Microbes 2017, 8, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Dang, N.; Ren, D.; Zhao, F.; Lv, R.; Ma, T.; Bao, Q.; Menghe, B.; Liu, W. Comparison of Bacterial Microbiota in Raw Mare’s Milk and Koumiss Using PacBio Single Molecule Real-Time Sequencing Technology. Front. Microbiol. 2020, 11, 581610. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Whelan, F.J.; Herath, I.; Lee, C.H.; Collins, S.M.; Bercik, P.; Surette, M.G. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 2016, 8, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeClere, M.; Costa, M.C. Fecal microbiota in horses with asthma. J. Veter. Intern. Med. 2020, 34, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collinet, A.; Grimm, P.; Julliand, S.; Julliand, V. Multidimensional Approach for Investigating the Effects of an Antibiotic–Probiotic Combination on the Equine Hindgut Ecosystem and Microbial Fibrolysis. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Harlow, B.E.; Lawrence, L.M.; Flythe, M.D. Diarrhea-associated pathogens, lactobacilli and cellulolytic bacteria in equine feces: Responses to antibiotic challenge. Veter. Microbiol. 2013, 166, 225–232. [Google Scholar] [CrossRef]
- Freccero, F.; Lanci, A.; Mariella, J.; Viciani, E.; Quercia, S.; Castagnetti, A.; Castagnetti, C. Changes in the Fecal Microbiota Associated with a Broad-Spectrum Antimicrobial Administration in Hospitalized Neonatal Foals with Probiotics Supplementation. Animals 2021, 11, 2283. [Google Scholar] [CrossRef]
- Cheung, S.G.; Goldenthal, A.R.; Uhlemann, A.-C.; Mann, J.J.; Miller, J.M.; Sublette, M.E. Systematic Review of Gut Microbiota and Major Depression. Front. Psychiatry 2019, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Vázquez, L.; Riba, G.V.G.; Arija, V.; Canals, J. Composition of Gut Microbiota in Children with Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, M.; Silva, G.; Ramos, R.; Staempfli, H.; Arroyo, L.; Kim, P.; Weese, J. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments in horses. Veter. J. 2015, 205, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Morotomi, M.; Nagai, F.; Sakon, H.; Tanaka, R. Paraprevotella clara gen. nov., sp. nov. and Paraprevotella xylaniphila sp. nov., members of the family ‘Prevotellaceae’ isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2009, 59, 1895–1900. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PRE TMS | POST TMS | ||||
|---|---|---|---|---|---|
| PacBio | Illumina | PacBio | Illumina | Correlation (p-Value) | |
| Good-quality reads | 176,967 | 687,081 | 124,796 | 737,802 | |
| Observed genera | 224.22 (57.36) | 68.43 (9.22) | 169.11 (40.83) | 53.40 (5.58) | 0.4222 (0.081) |
| Chao | 354.27 (86.95) | 87.26 (14.74) | 253.45 (51.57) | 67.99 (5.29) | 0.3576 (0.145) |
| Simpson’s | 0.93 (0.08) | 5.68 (0.98) | 0.93 (0.04) | 4.44 (1.93) | 0.0724 (0.775) |
| Shannon | 5.56 (0.99) | 2.23 (0.17) | 4.90 (0.73) | 1.97 (0.37) | 0.4519 (0.060) |
| PacBio | Illumina | |
|---|---|---|
| Observed genera | 0.651 | 0.004 * |
| Chao | 0.033 * | 0.005 * |
| Simpson’s | 1.000 | 0.197 |
| Shannon | 0.149 | 0.148 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pietro, R.; Arroyo, L.G.; Leclere, M.; Costa, M.C. Species-Level Gut Microbiota Analysis after Antibiotic-Induced Dysbiosis in Horses. Animals 2021, 11, 2859. https://doi.org/10.3390/ani11102859
Di Pietro R, Arroyo LG, Leclere M, Costa MC. Species-Level Gut Microbiota Analysis after Antibiotic-Induced Dysbiosis in Horses. Animals. 2021; 11(10):2859. https://doi.org/10.3390/ani11102859
Chicago/Turabian StyleDi Pietro, Rebecca, Luis G. Arroyo, Mathilde Leclere, and Marcio Carvalho Costa. 2021. "Species-Level Gut Microbiota Analysis after Antibiotic-Induced Dysbiosis in Horses" Animals 11, no. 10: 2859. https://doi.org/10.3390/ani11102859
APA StyleDi Pietro, R., Arroyo, L. G., Leclere, M., & Costa, M. C. (2021). Species-Level Gut Microbiota Analysis after Antibiotic-Induced Dysbiosis in Horses. Animals, 11(10), 2859. https://doi.org/10.3390/ani11102859