Identification of Target Chicken Populations by Machine Learning Models Using the Minimum Number of SNPs

 ,
,  ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. DNA Extraction
2.3. High-Density SNP Genotyping and Quality Control (QC)
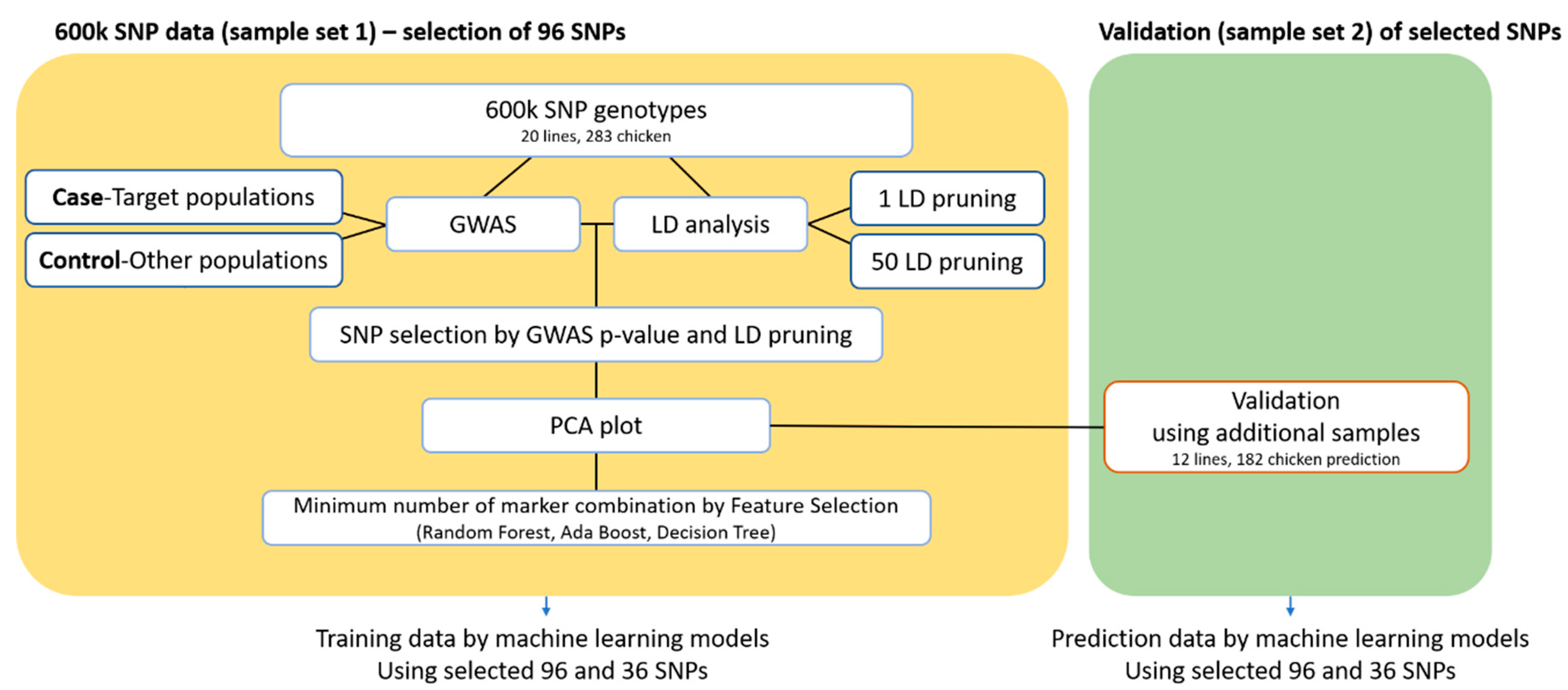
2.4. Selection of 96 Candidate SNP Markers for Identification of the Target Population
2.5. Machine Learning Approach for Determining the Combination with the Minimum Number of Markers Required for Breed Identification
3. Results
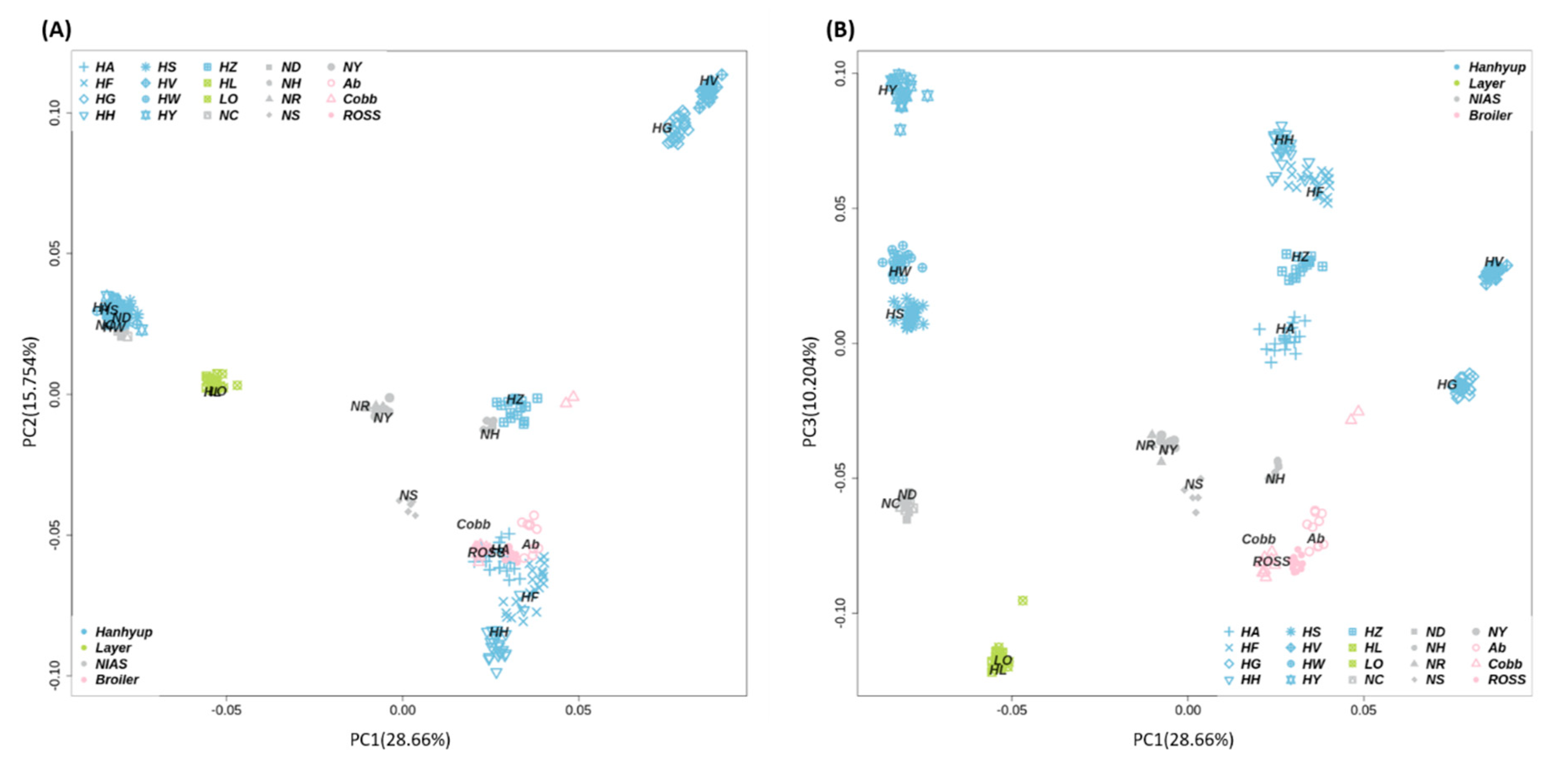
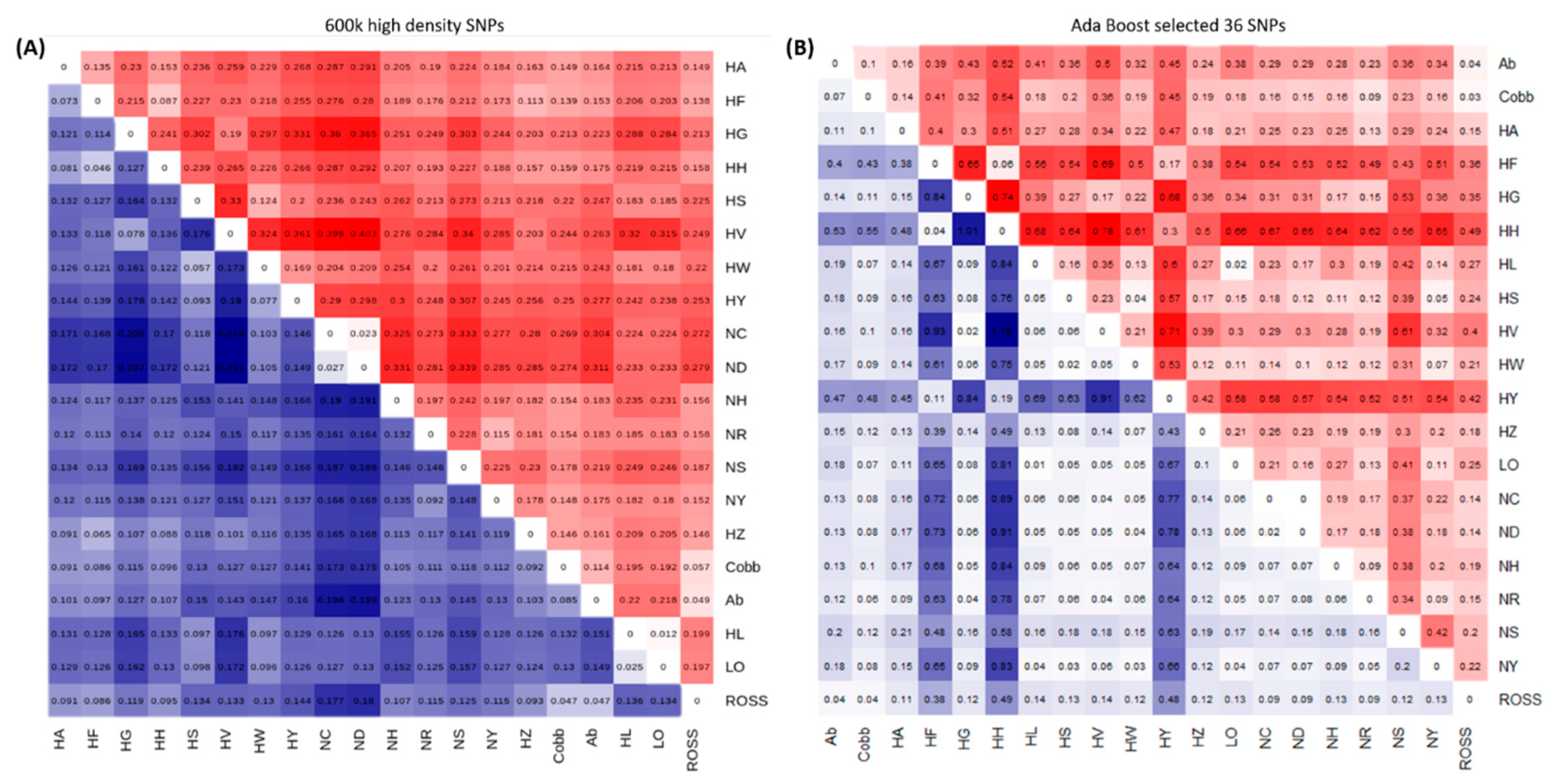
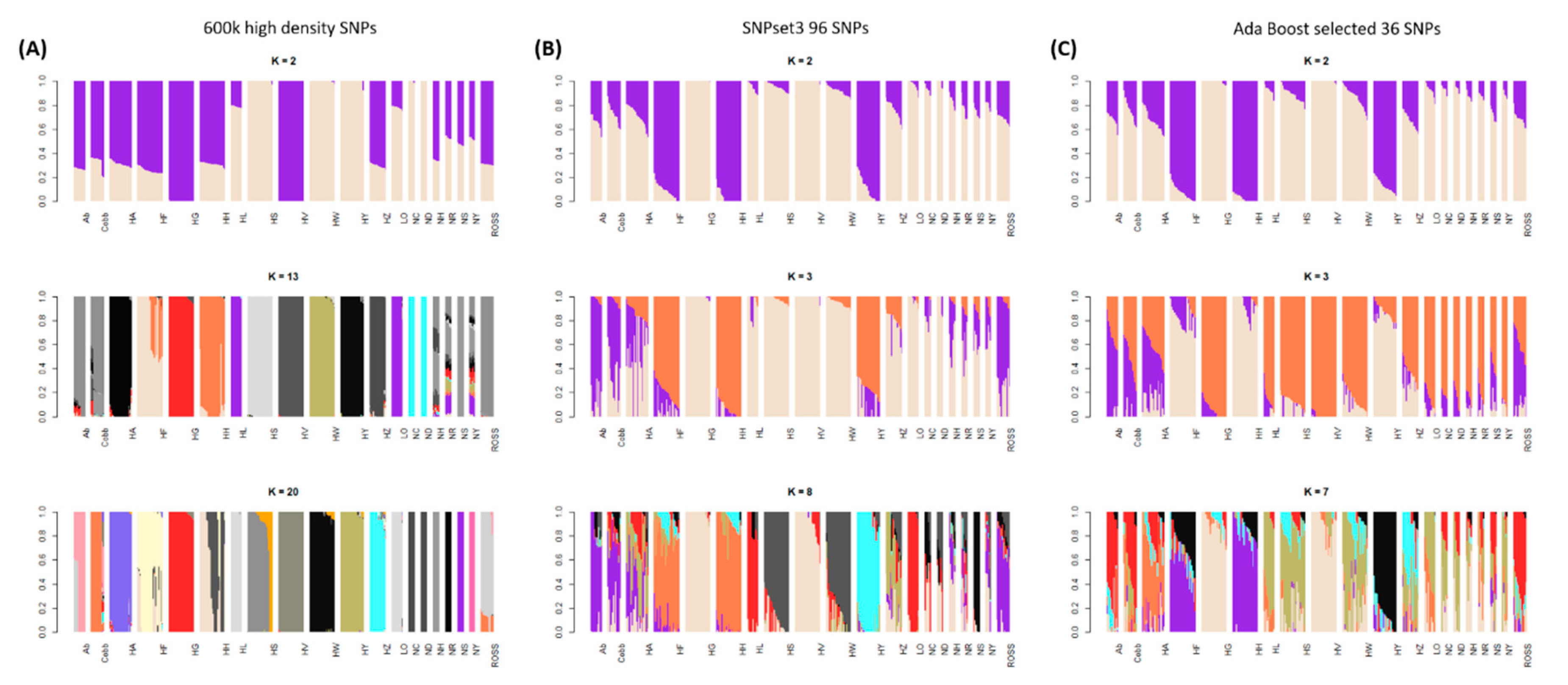
3.1. Genetic Diversity Analyses to Identify SNP Marker Combinations
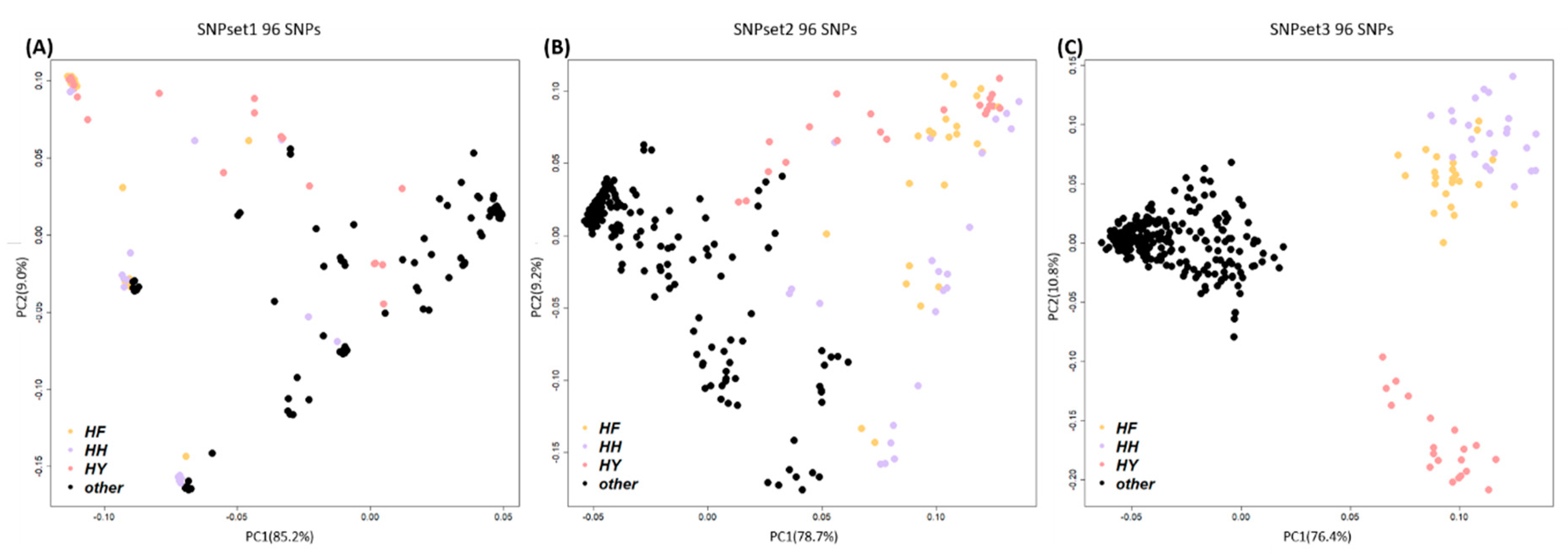
3.2. GWAS and SNP Selection
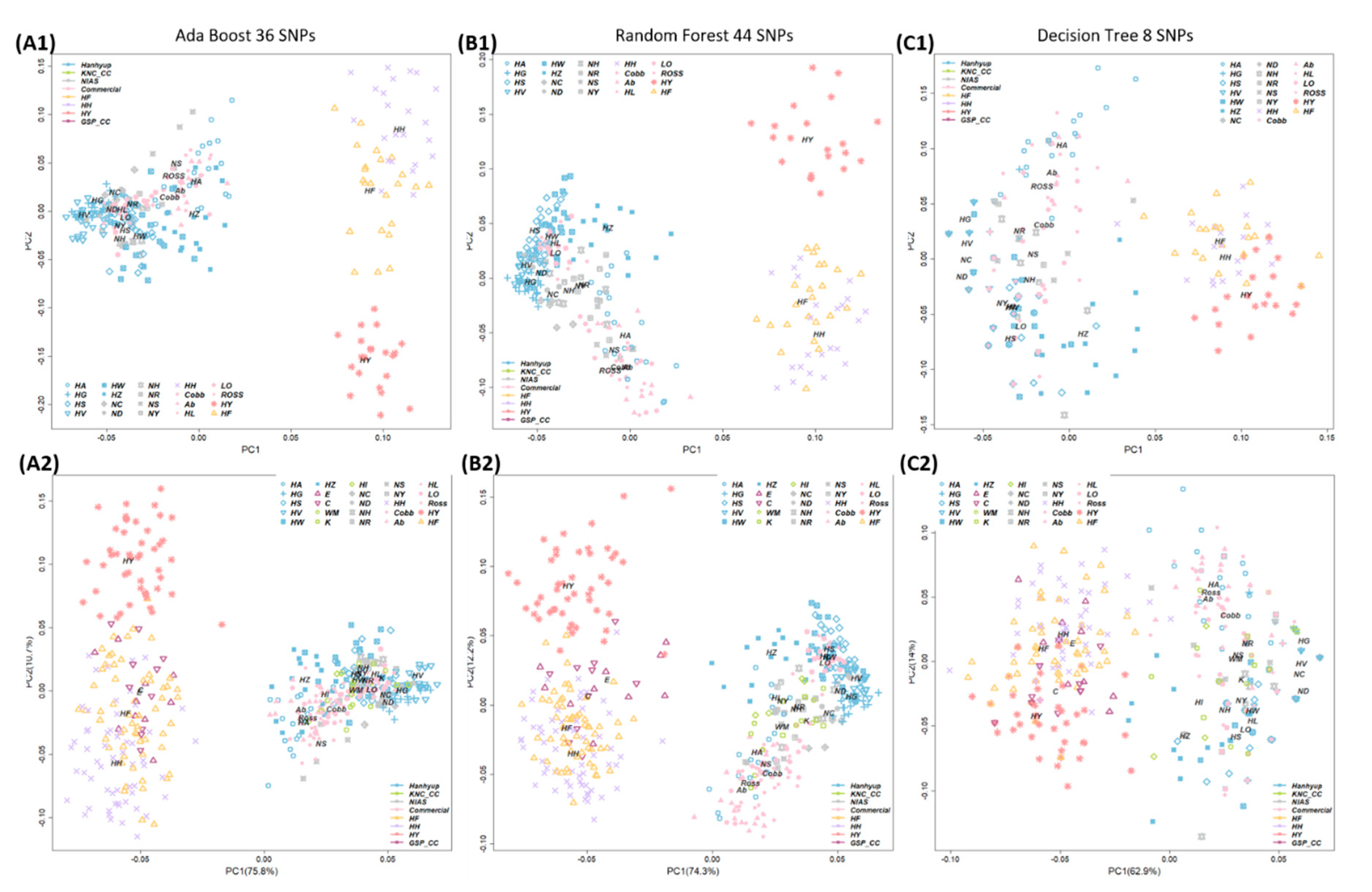
3.3. Breed Identification by Machine-Learning Algorithms Using the Minimum Number of SNPs
3.4. Validation Study Using Additional Samples
4. Discussion
4.1. Identification of Target Chicken Population Based on Genetic Components
4.2. GWAS and LD Analysis for Identification of the Target Population
4.3. Machine Learning Algorithms for Classification of the Case and Control Chicken Populations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yeung, R.M.; Morris, J. Consumer perception of food risk in chicken meat. Nutr. Food Sci. 2001. [Google Scholar] [CrossRef]
- MAFRA (Ministry of Agriculture, Food and Rural Affairs). Major Statistics of the Ministry of Agriculture, Food and Rural Affairs 2019. Available online: http://library.mafra.go.kr/skyblueimage/28195.pdf (accessed on 1 November 2020).
- Shim, J.-M.; Seo, D.-W.; Seo, S.; Kim, J.-J.; Min, D.-M.; Kim, J.; Jeon, J.-T.; Lee, J.-H. Discrimination of Korean cattle (Hanwoo) with imported beef from USA based on the SNP markers. Korean J. Food Sci. Anim. Resour. 2010, 30, 918–922. [Google Scholar] [CrossRef]
- Oh, J.-D.; Song, K.-D.; Seo, J.-H.; Kim, D.-K.; Kim, S.-H.; Seo, K.-S.; Lim, H.-T.; Lee, J.-B.; Park, H.-C.; Ryu, Y.-C. Genetic traceability of black pig meats using microsatellite markers. Asian Australas. J. Anim. Sci. 2014, 27, 926. [Google Scholar] [CrossRef][Green Version]
- Kim, K.; Seo, M.; Kang, H.; Cho, S.; Kim, H.; Seo, K.-S. Application of logitboost classifier for traceability using snp chip data. PLoS ONE 2015, 10, e0139685. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.-R.; Hoque, M.R.; Seo, D.-W.; Sultana, H.; Park, H.-B.; Lim, H.-T.; Heo, K.-N.; Kang, B.-S.; Jo, C.; Lee, J.-H. ISAG-recommended Microsatellite Marker Analysis among Five Korean Native Chicken Lines. J. Anim. Sci. Technol. 2012, 54, 401–409. [Google Scholar] [CrossRef]
- Dalvit, C.; De Marchi, M.; Cassandro, M. Genetic traceability of livestock products: A review. Meat Sci. 2007, 77, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Burt, D.W. Chicken genome: Current status and future opportunities. Genome Res. 2005, 15, 1692–1698. [Google Scholar] [CrossRef]
- Hillier, L.W.; Miller, W.; Birney, E.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.A.; Delany, M.E. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2014, 423, 695–777. [Google Scholar] [CrossRef]
- Groenen, M.A.; Megens, H.-J.; Zare, Y.; Warren, W.C.; Hillier, L.W.; Crooijmans, R.P.; Vereijken, A.; Okimoto, R.; Muir, W.M.; Cheng, H.H. The development and characterization of a 60K SNP chip for chicken. BMC Genom. 2011, 12, 274. [Google Scholar] [CrossRef]
- Kranis, A.; Gheyas, A.A.; Boschiero, C.; Turner, F.; Yu, L.; Smith, S.; Talbot, R.; Pirani, A.; Brew, F.; Kaiser, P.; et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genom. 2013, 14, 59. [Google Scholar] [CrossRef]
- Karniol, B.; Shirak, A.; Baruch, E.; Singrün, C.; Tal, A.; Cahana, A.; Kam, M.; Skalski, Y.; Brem, G.; Weller, J. Development of a 25-plex SNP assay for traceability in cattle. Anim. Genet. 2009, 40, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Bourbon, M.; Williams, M.; Humphries, S.E. Clinical utility of the polygenic LDL-C SNP score in familial hypercholesterolemia. Atherosclerosis 2018, 277, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Vignal, A.; Milan, D.; SanCristobal, M.; Eggen, A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet. Sel. Evol. 2002, 34, 275–305. [Google Scholar] [CrossRef] [PubMed]
- Suekawa, Y.; Aihara, H.; Araki, M.; Hosokawa, D.; Mannen, H.; Sasazaki, S. Development of breed identification markers based on a bovine 50K SNP array. Meat Sci. 2010, 85, 285–288. [Google Scholar] [CrossRef]
- Brooks, A.; Creighton, E.K.; Gandolfi, B.; Khan, R.; Grahn, R.A.; Lyons, L.A. SNP Miniplexes for Individual Identification of Random-Bred Domestic Cats. J. Forensic Sci. 2016, 61, 594–606. [Google Scholar] [CrossRef]
- Kumar, H.; Panigrahi, M.; Chhotaray, S.; Parida, S.; Chauhan, A.; Bhushan, B.; Gaur, G.K.; Mishra, B.P.; Singh, R.K. Comparative analysis of five different methods to design a breed-specific SNP panel for cattle. Anim. Biotechnol. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Mitchell, T.M. Machine learning and data mining. Commun. ACM 1999, 42, 30–36. [Google Scholar] [CrossRef]
- Guinand, B.; Topchy, A.; Page, K.; Burnham-Curtis, M.; Punch, W.; Scribner, K. Comparisons of likelihood and machine learning methods of individual classification. J. Hered. 2002, 93, 260–269. [Google Scholar] [CrossRef]
- Bertolini, F.; Galimberti, G.; Calò, D.; Schiavo, G.; Matassino, D.; Fontanesi, L. Combined use of principal component analysis and random forests identify population-informative single nucleotide polymorphisms: Application in cattle breeds. J. Anim. Breed. Genet. 2015, 132, 346–356. [Google Scholar] [CrossRef]
- Bertolini, F.; Galimberti, G.; Schiavo, G.; Mastrangelo, S.; Di Gerlando, R.; Strillacci, M.; Bagnato, A.; Portolano, B.; Fontanesi, L. Preselection statistics and Random Forest classification identify population informative single nucleotide polymorphisms in cosmopolitan and autochthonous cattle breeds. Animal 2017, 12, 12–19. [Google Scholar] [CrossRef]
- Pasupa, K.; Rathasamuth, W.; Tongsima, S. Discovery of significant porcine SNPs for swine breed identification by a hybrid of information gain, genetic algorithm, and frequency feature selection technique. BMC Bioinform. 2020, 21, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Bertolini, F.; Galimberti, G.; Bovo, S.; Dall’Olio, S.; Costa, L.N.; Gallo, M.; Fontanesi, L. A machine learning approach for the identification of population-informative markers from high-throughput genotyping data: Application to several pig breeds. Animal 2020, 14, 223–232. [Google Scholar] [CrossRef]
- Judge, M.; Kelleher, M.; Kearney, J.; Sleator, R.; Berry, D. Ultra-low-density genotype panels for breed assignment of Angus and Hereford cattle. Animal 2017, 11, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Koo, B.; Kim, E.; Heo, J.M. Comparison of growth performance between crossbred Korean native chickens for hatch to 28 days. CNU J. Agric. Sci. 2015, 42, 23–27. [Google Scholar] [CrossRef][Green Version]
- Jin, S.; Jayasena, D.; Jo, C.; Lee, J. The breeding history and commercial development of the Korean native chicken. World’s Poult. Sci. J. 2017, 73, 163–174. [Google Scholar] [CrossRef]
- Seo, D.; Lee, D.H.; Choi, N.; Sudrajad, P.; Lee, S.-H.; Lee, J.-H. Estimation of linkage disequilibrium and analysis of genetic diversity in Korean chicken lines. PLoS ONE 2018, 13, e0192063. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Genetic Distance between Populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. ESTIMATING F-STATISTICS FOR THE ANALYSIS OF POPULATION STRUCTURE. Int. J. Org. Evol. 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013; Available online: https://www.R-project.org/.2015.02.10 (accessed on 1 November 2020).
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. Available online: http://www.math.chalmers.se/Stat/Grundutb/GU/MSA220/S18/caret-JSS.pdf.2008.11.10 (accessed on 1 November 2020). [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kégl, B. The return of AdaBoost. MH: Multi-class Hamming trees. arXiv 2013, arXiv:1312.6086. [Google Scholar]
- Singh, A.; Thakur, N.; Sharma, A. A Review of Supervised Machine Learning Algorithms. In Proceedings of the 2016 3rd International Conference on Computing for Sustainable Global Development (INDIACom), New Delhi, India, 16–18 March 2016; pp. 1310–1315. Available online: https://ieeexplore.ieee.org/abstract/document/7724478.2016.03.16 (accessed on 1 November 2020).
- Tharwat, A. Linear vs. quadratic discriminant analysis classifier: A tutorial. Int. J. Appl. Pattern Recognit. 2016, 3, 145–180. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Diagnostic tests. 1: Sensitivity and specificity. BMJ Br. Med. J. 1994, 308, 1552. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, C.; Wang, X.; Li, Z.; Sun, G.; Li, G.; Liu, X.; Kang, X.; Han, R. Genetic diversity of mtDNA D-loop sequences in four native Chinese chicken breeds. Br. Poult. Sci. 2017, 58, 490–497. [Google Scholar] [CrossRef]
- Dimauro, C.; Cellesi, M.; Steri, R.; Gaspa, G.; Sorbolini, S.; Stella, A.; Macciotta, N.P.P. Use of the canonical discriminant analysis to select SNP markers for bovine breed assignment and traceability purposes. Anim. Genet. 2013, 44, 377–382. [Google Scholar] [CrossRef]
- Pérez-Enciso, M.; Zingaretti, L.M. A guide on deep learning for complex trait genomic prediction. Genes 2019, 10, 553. [Google Scholar] [CrossRef]
- Alves, A.A.C.; da Costa, R.M.; Bresolin, T.; Fernandes Júnior, G.A.; Espigolan, R.; Ribeiro, A.M.F.; Carvalheiro, R.; Albuquerque, L.G.d. Genome-wide prediction for complex traits under the presence of dominance effects in simulated populations using GBLUP and machine learning methods. J. Anim. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, M.L.; Pong-Wong, R.; Spiliopoulou, A.; Hayward, C.; Rudan, I.; Campbell, H.; Wright, A.F.; Wilson, J.F.; Agakov, F.; Navarro, P. Application of high-dimensional feature selection: Evaluation for genomic prediction in man. Sci. Rep. 2015, 5, 10312. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.M.; Megens, H.J.; Crooijmans, R.P.M.A.; Schook, L.B.; Groenen, M.A.M. Identification of High Utility SNPs for Population Assignment and Traceability Purposes in the Pig Using High-throughput Sequencing. Anim. Genet. 2011, 42, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Ciampolini, R.; Cecchi, F.; Spinetti, I.; Rocchi, A.; Biscarini, F. The Use of Genetic Markers to Estimate Relationships between Dogs in the Course of Criminal Investigations. BMC Res. Notes 2017, 10, 414. [Google Scholar] [CrossRef]
- Carroll, E.L.; Bruford, M.W.; DeWoody, J.A.; Leroy, G.; Strand, A.; Waits, L.; Wang, J. Genetic and Genomic Monitoring with Minimally Invasive Sampling Methods. Evol. Appl. 2018, 11, 1094–1119. [Google Scholar] [CrossRef]
- Biscarini, F.; Marini, S.; Stevanato, P.; Broccanello, C.; Bellazzi, R.; Nazzicari, N. Developing a parsimonius predictor for binary traits in sugar beet (Beta vulgaris). Mol. Breed. 2015, 35, 10. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chicken Group | Population Code | Origin of Population | Description | 600 k Array (Sample Set 1) | Validation (Sample Set 2) |
|---|---|---|---|---|---|
| Government-maintained chicken (NIAS) | NC | Rhode Island Red | Imported (1960s) and locally adapted chicken population | 6 | |
| ND | 6 | ||||
| NH | Cornish | 6 | |||
| NS | 6 | ||||
| NR | Red-brown Korean native chicken | Purebred Korean native chicken | 6 | ||
| NY | Yellow-brown Korean native chicken | 5 | |||
| Commercial native chicken | HH | Hanhyup Farm | Founder population for new chicken breeding stock | 23 | 36 |
| HF | 23 | 36 | |||
| HY | 21 | 26 | |||
| HW | Maintained population | 23 | |||
| HS | 23 | ||||
| HG | 23 | ||||
| HV | 23 | ||||
| HA | 20 | ||||
| HZ | 15 | ||||
| 1E | HH, HF, HY cross | 10 | |||
| 2C | HH, HF, HY cross | 10 | |||
| WM_2 | Woorimatdaq ver2 | NIAS-developed crossed chicken population | 10 | ||
| Yelim K | Yelim Farm | Private population | 5 | ||
| HI | Hyunin Farm | Private population | 5 | ||
| Commercial broiler | Ab | Arbor Acre | Meat-type chicken | 10 | 11 |
| Cobb | Cobb broiler | 12 | 8 | ||
| Ross | Ross broiler | 12 | 20 | ||
| Commercial layer | LO | Lohmann brown | Egg-producing chicken | 10 | 5 |
| HL | Hyline brown | 10 | |||
| Total | 283 | 182 | |||
| Classification Model | Accuracy | 1 AUC | Precision | Sensitivity (2 TPR) | Specificity (3 TNR) |
|---|---|---|---|---|---|
| AdaBoost: 33 selected markers | |||||
| Decision Tree | 0.995 | 0.996 | 1 | 0.992 | 1 |
| AdaBoost | 0.995 | 0.996 | 1 | 0.992 | 1 |
| Linear 4 SVM | 0.995 | 0.996 | 1 | 0.992 | 1 |
| 5 QDA | 0.995 | 0.996 | 1 | 0.992 | 1 |
| Random Forest | 0.995 | 0.996 | 1 | 0.992 | 1 |
| 6 LDA | 1 | 1 | 1 | 1 | 1 |
| K-Nearest Neighbors | 1 | 1 | 1 | 1 | 1 |
| Naïve Bayes | 0.995 | 0.996 | 1 | 0.992 | 1 |
| Random Forest: 44 selected markers | |||||
| Decision Tree | 0.969 | 0.975 | 1 | 0.949 | 1 |
| AdaBoost | 0.969 | 0.975 | 1 | 0.949 | 1 |
| Linear SVM | 0.990 | 0.992 | 1 | 0.983 | 1 |
| QDA | 0.995 | 0.996 | 1 | 0.992 | 1 |
| Random Forest | 0.969 | 0.975 | 1 | 0.949 | 1 |
| LDA | 1 | 1 | 1 | 1 | 1 |
| K-Nearest Neighbors | 0.984 | 0.987 | 1 | 0.975 | 1 |
| Naïve Bayes | 0.969 | 0.975 | 1 | 0.949 | 1 |
| Decision Tree: 8 selected markers | |||||
| Decision Tree | 0.984 | 0.987 | 1 | 0.975 | 1 |
| AdaBoost | 0.984 | 0.987 | 1 | 0.975 | 1 |
| Linear SVM | 0.964 | 0.970 | 1 | 0.941 | 1 |
| QDA | 0.974 | 0.979 | 1 | 0.958 | 1 |
| Random Forest | 0.979 | 0.981 | 0.991 | 0.975 | 0.986 |
| LDA | 0.995 | 0.996 | 1 | 0.992 | 1 |
| K-Nearest Neighbors | 0.984 | 0.987 | 1 | 0.975 | 1 |
| Naïve Bayes | 0.969 | 0.975 | 1 | 0.949 | 1 |
| Pop | N | AdaBoost | Random Forest | Decision Tree | Linear DiscriminantAnalysis | Naïve Bayes | Nearest Neighbor | Quadratic Discriminant Analysis |
|---|---|---|---|---|---|---|---|---|
| HH | 36 | 1 | 1 | 0.972 | 1 | 1 | 0.972 | 0.972 |
| HF | 36 | 1 | 1 | 0.972 | 1 | 1 | 1 | 1 |
| HY | 26 | 0.962 | 0.885 | 0.769 | 0.962 | 1 | 0.962 | 0.923 |
| 1E | 10 | 1 | 0.7 | 0.9 | 1 | 1 | 1 | 1 |
| 2C | 10 | 1 | 1 | 0.8 | 1 | 1 | 1 | 0.9 |
| Ab | 11 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Cobb | 8 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Ross | 20 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| LO | 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| WM_2 | 10 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Yelim K | 5 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| HI | 5 | 1 | 11 | 1 | 1 | 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, D.; Cho, S.; Manjula, P.; Choi, N.; Kim, Y.-K.; Koh, Y.J.; Lee, S.H.; Kim, H.-Y.; Lee, J.H. Identification of Target Chicken Populations by Machine Learning Models Using the Minimum Number of SNPs. Animals 2021, 11, 241. https://doi.org/10.3390/ani11010241
Seo D, Cho S, Manjula P, Choi N, Kim Y-K, Koh YJ, Lee SH, Kim H-Y, Lee JH. Identification of Target Chicken Populations by Machine Learning Models Using the Minimum Number of SNPs. Animals. 2021; 11(1):241. https://doi.org/10.3390/ani11010241
Chicago/Turabian StyleSeo, Dongwon, Sunghyun Cho, Prabuddha Manjula, Nuri Choi, Young-Kuk Kim, Yeong Jun Koh, Seung Hwan Lee, Hyung-Yong Kim, and Jun Heon Lee. 2021. "Identification of Target Chicken Populations by Machine Learning Models Using the Minimum Number of SNPs" Animals 11, no. 1: 241. https://doi.org/10.3390/ani11010241
APA StyleSeo, D., Cho, S., Manjula, P., Choi, N., Kim, Y.-K., Koh, Y. J., Lee, S. H., Kim, H.-Y., & Lee, J. H. (2021). Identification of Target Chicken Populations by Machine Learning Models Using the Minimum Number of SNPs. Animals, 11(1), 241. https://doi.org/10.3390/ani11010241