Sex Differences in Intestinal Microbial Composition and Function of Hainan Special Wild Boar

 and
and 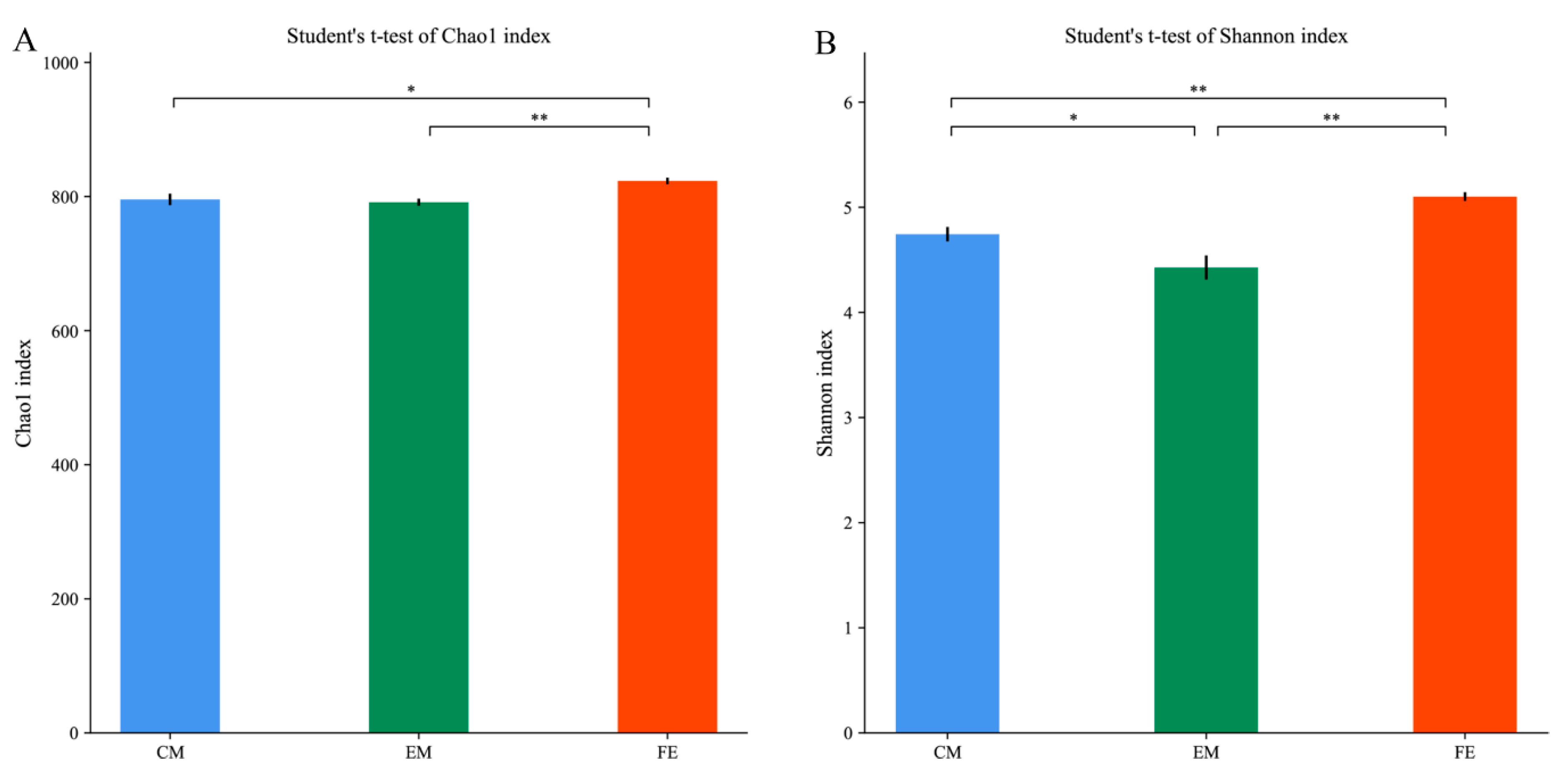{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Sample Collection
2.2. DNA Extraction and PCR Amplification
2.3. 16S rRNA Gene Sequencing and Sequence Splicing
2.4. Species Annotation and Taxonomic Analysis
2.5. Diversity Analysis and Significant Difference Analysis between Groups
2.6. Predictive Analysis of Functional Genes
3. Results
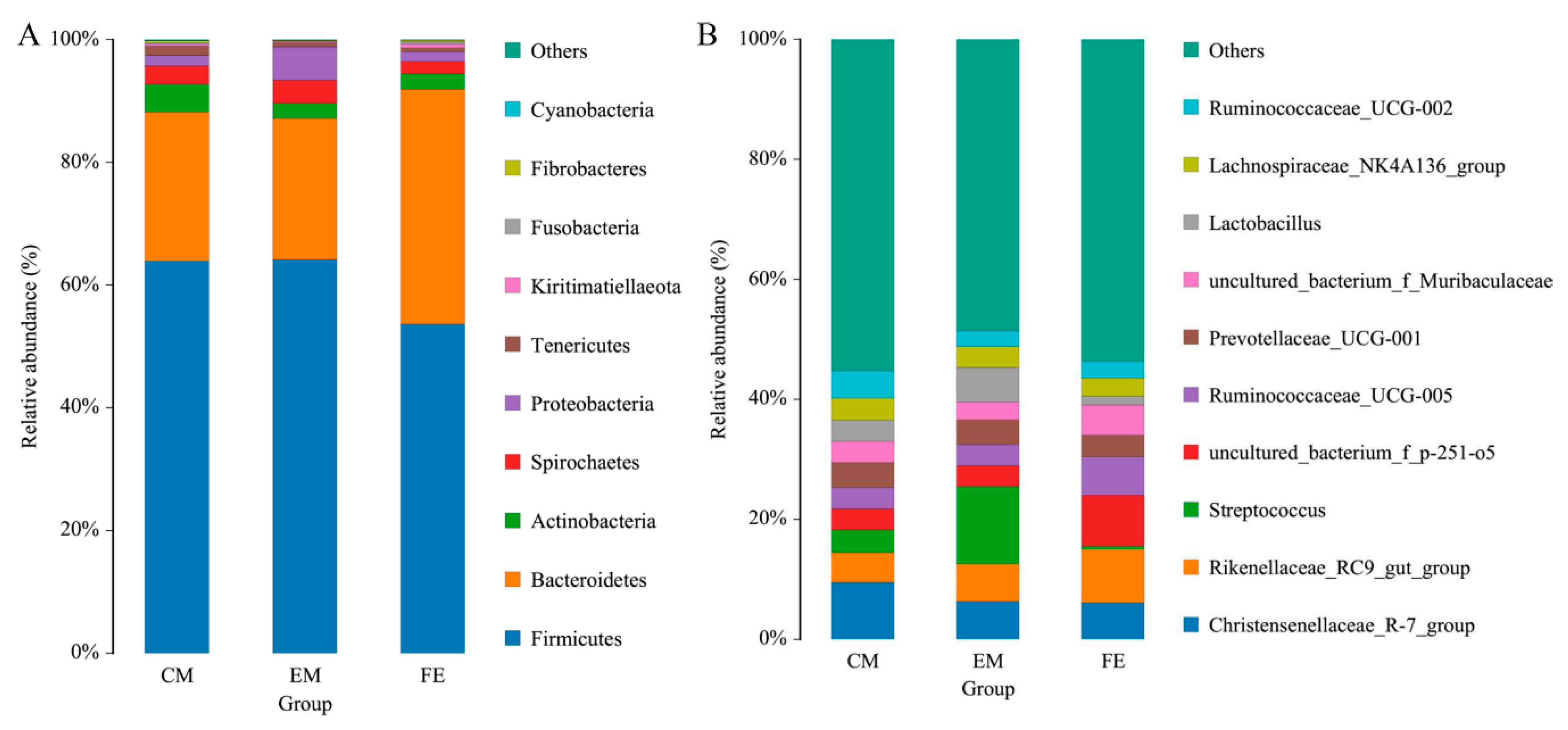
3.1. OTU Clustering and Species Taxonomy Analysis
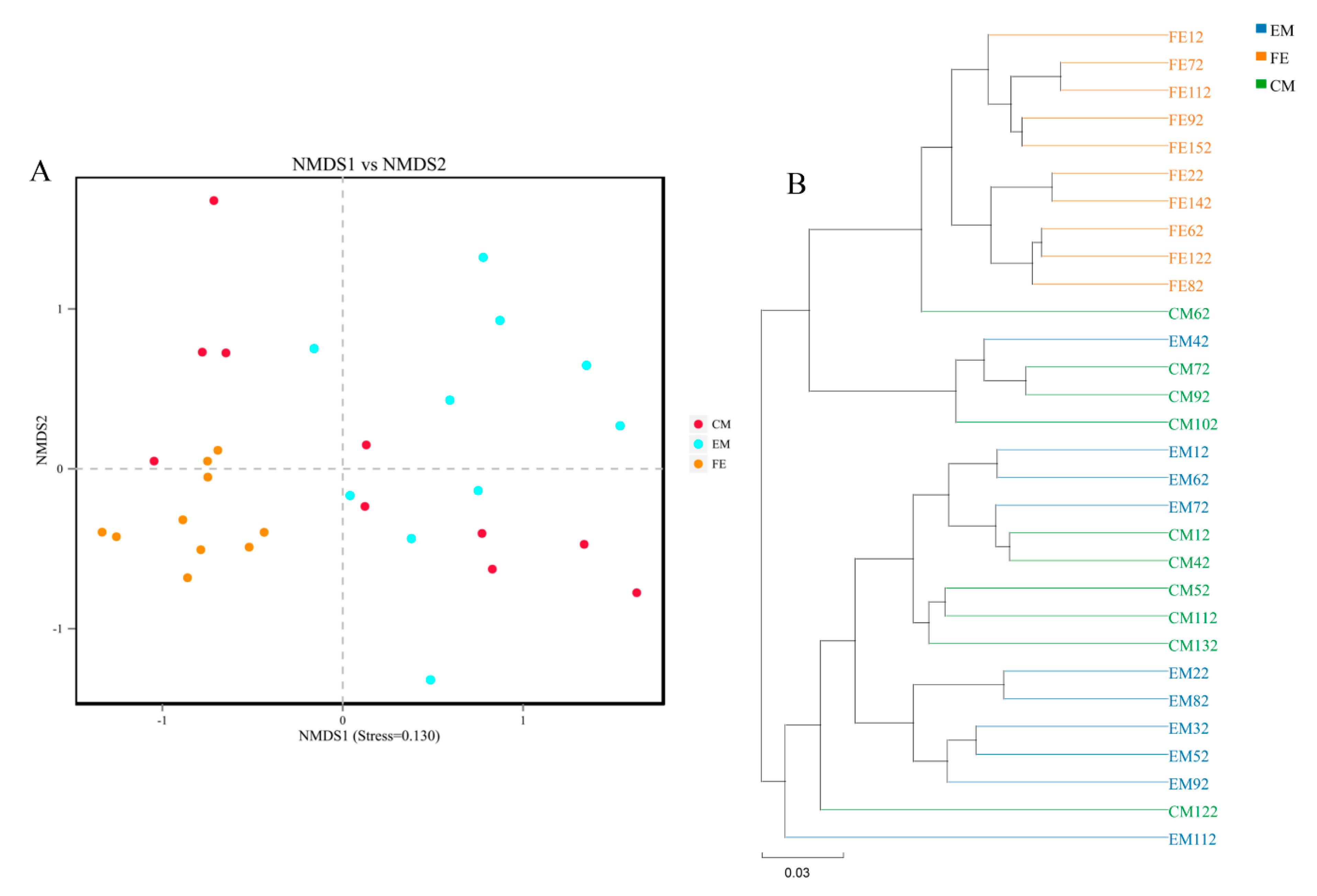
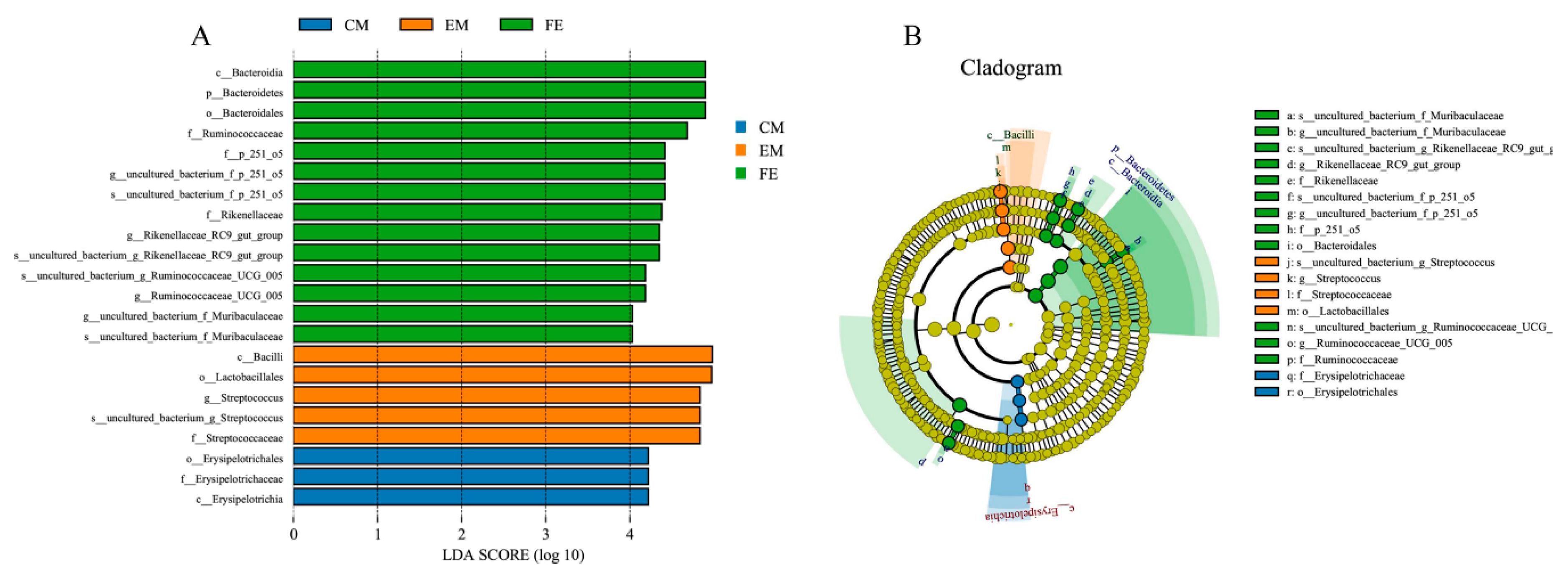
3.2. Comparison of Fecal Microorganisms among Three Groups of Pigs
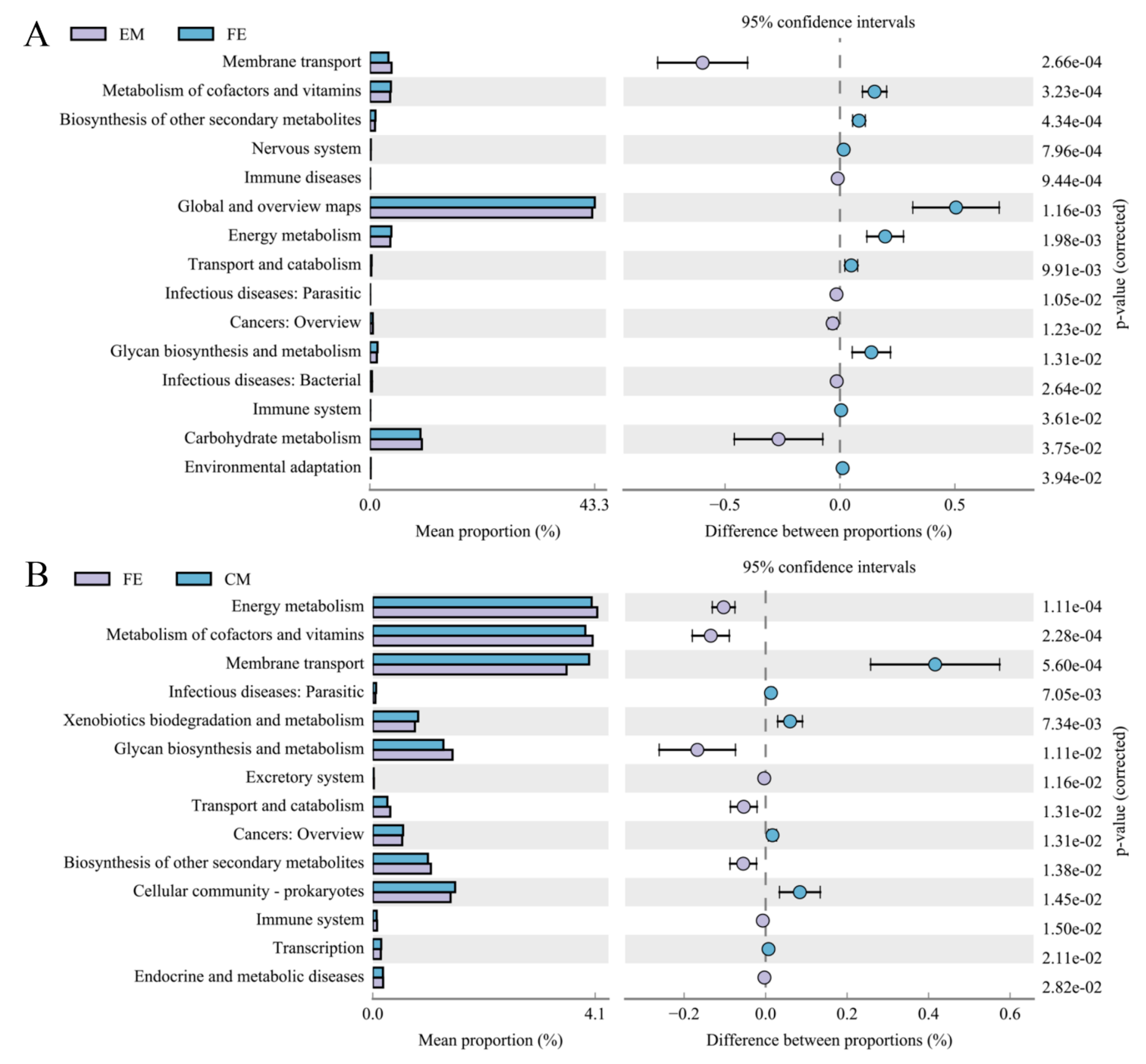
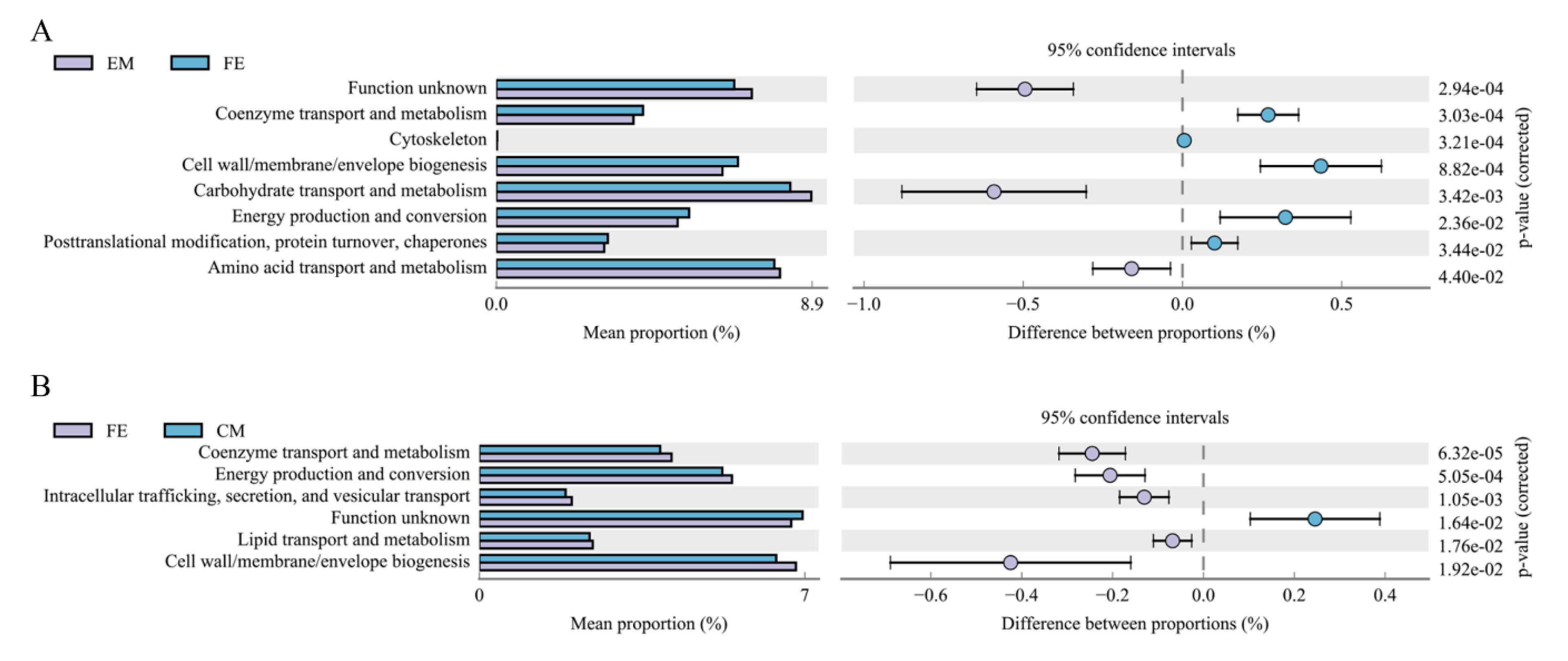
3.3. Differences of Microbial Function among Three Groups of Pigs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Org, E.; Parks, B.W.; Joo, J.W.; Emert, B.; Schwartzman, W.; Kang, E.Y.; Mehrabian, M.; Pan, C.; Knight, R.; Gunsalus, R.; et al. Genetic and environmental control of host-gut microbiota interactions. Genome Res. 2015, 25, 1558–1569. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.P.; Kong, F.L.; Xiang, Y.; Zhou, W.D.; Wang, J.J.; Yang, H.; Zhang, G.L.; Zhao, J.C. Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci. Rep. UK 2018, 8, 5985. [Google Scholar] [CrossRef]
- Kovacs, A.; Ben-Jacob, N.; Tayem, H.; Halperin, E.; Iraqi, F.A.; Gophna, U. Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb. Ecol. 2011, 61, 423–428. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Burrows, M.; Khan, A.A.; Graham, L.; Volchkov, P.; Becker, L.; Antonopoulos, D.; Umesaki, Y.; Chervonsky, A.V. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013, 39, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.M.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex Differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef]
- Org, E.; Mehrabian, M.; Parks, B.W.; Shipkova, P.; Liu, X.Q.; Drake, T.A.; Lusis, A.J. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016, 7, 313–322. [Google Scholar] [CrossRef]
- Fushuku, S.; Fukuda, K. Gender difference in the composition of fecal flora in laboratory mice, as detected by denaturing gradient gel electrophoresis (DGGE). Exp. Anim. 2008, 57, 489–493. [Google Scholar] [CrossRef]
- Elderman, M.; Hugenholtz, F.; Belzer, C.; Boekschoten, M.; van Beek, A.; de Haan, B.; Savelkoul, H.; de Vos, P.; Faas, M. Sex and strain dependent differences in mucosal immunology and microbiota composition in mice. Biol. Sex Differ. 2018, 9, 26. [Google Scholar] [CrossRef]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Meijer, B.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.; de Jonge, M.I.; Faas, M.M.; Boekschoten, M.V.; et al. The impact of gut microbiota on gender-specific differences in immunity. Front. Immunol. 2017, 8, 754. [Google Scholar] [CrossRef] [PubMed]
- Dicksved, J.; Floistrup, H.; Bergstrom, A.; Rosenquist, M.; Pershagen, G.; Scheynius, A.; Roos, S.; Alm, J.S.; Engstrand, L.; Braun-Fahrlander, C.; et al. Molecular fingerprinting of the fecal microbiota of children raised according to different lifestyles. Appl. Environ. Microbiol. 2007, 73, 2284–2289. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: A cross-sectional study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Bolnick, D.I.; Snowberg, L.K.; Hirsch, P.E.; Lauber, C.L.; Org, E.; Parks, B.; Lusis, A.J.; Knight, R.; Caporaso, J.G.; Svanback, R. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Tetel, M.J.; de Vries, G.J.; Melcangi, R.C.; Panzica, G.; O‘Mahony, S.M. Steroids, stress and the gut microbiome-brain axis. J. Neuroendocrinol. 2018, 30. [Google Scholar] [CrossRef]
- Harada, N.; Hanaoka, R.; Horiuchi, H.; Kitakaze, T.; Mitani, T.; Inui, H.; Yamaji, R. Castration influences intestinal microflora and induces abdominal obesity in high-fat diet-fed mice. Sci. Rep. UK 2016, 6. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Zhao, J.; Ding, X.; Men, G. Sex differences in resting state network (RSN) functional connections with mild cognitive impairment (MCI) progression. Neurosci. Lett. 2020, 724, 134891. [Google Scholar] [CrossRef]
- He, M.Z.; Gao, J.; Wu, J.Y.; Zhou, Y.Y.; Fu, H.; Ke, S.L.; Yang, H.; Chen, C.Y.; Huang, L.S. Host gender and androgen levels regulate gut bacterial taxa in pigs leading to sex-biased serum metabolite profiles. Front. Microbiol. 2019, 10, 1359. [Google Scholar] [CrossRef]
- Zhou, Z.J.; Zheng, W.J.; Shang, W.W.; Du, H.L.; Li, G.L.; Yao, W. How host gender affects the bacterial community in pig feces and its correlation to skatole production. Ann. Microbiol. 2015, 65, 2379–2386. [Google Scholar] [CrossRef]
- Kong, F.L.; Zhao, J.C.; Han, S.S.; Zeng, B.; Yang, J.D.; Si, X.H.; Yang, B.Q.; Yang, M.Y.; Xu, H.L.; Li, Y. Characterization of the gut microbiota in the red panda (Ailurus fulgens). PLoS ONE 2014, 9, e87885. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef] [PubMed]
- Fogel, A.T. The gut microbiome of wild lemurs: A comparison of sympatric lemur catta and Propithecus verreauxi. Folia Primatol. Int. J. Primatol. 2015, 86, 85–95. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, W.; Wang, L.; Hou, R.; Zhang, M.; Fei, L.; Zhang, X.; Huang, H.; Bridgewater, L.C.; Jiang, Y.; et al. The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. mBio 2015, 6, e00022-15. [Google Scholar] [CrossRef]
- Zeng, Y.; Zeng, D.; Zhou, Y.; Niu, L.; Deng, J.; Li, Y.; Pu, Y.; Lin, Y.; Xu, S.; Liu, Q.; et al. Microbial biogeography along the gastrointestinal tract of a red panda. Front. Microbiol. 2018, 9, 1411. [Google Scholar] [CrossRef]
- Krishnan, S.; Alden, N.; Lee, K. Pathways and functions of gut microbiota metabolism impacting host physiology. Curr. Opin. Biotechnol. 2015, 36, 137–145. [Google Scholar] [CrossRef]
- Matsui, T.; Tanaka, J.; Namihira, T.; Shinzato, N. Antibiotics production by an actinomycete isolated from the termite gut. J. Basic Microbiol. 2012, 52, 731–735. [Google Scholar] [CrossRef]
- Tao, S.; Bai, Y.; Zhou, X.; Zhao, J.; Yang, H.; Zhang, S.; Wang, J. In vitro fermentation characteristics for different ratios of soluble to insoluble dietary fiber by fresh fecal microbiota from growing pigs. ACS Omega 2019, 4, 15158–15167. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Wu, Y.J.; Ye, H.; Feng, C.P.; Han, D.D.; Tao, S.Y.; Pi, Y.; Zhao, J.Y.; Chen, L.J.; Wang, J.J. Dietary milk fat globule membrane supplementation during late gestation increased the growth of neonatal piglets by improving their plasma parameters, intestinal barriers, and fecal microbiota. RSC Adv. 2020, 10, 16987–16998. [Google Scholar] [CrossRef]
- Reddy, K.E.; Kim, H.R.; Jeong, J.Y.; So, K.M.; Lee, S.; Ji, S.Y.; Kim, M.; Lee, H.J.; Lee, S.; Kim, K.H.; et al. Impact of breed on the fecal microbiome of dogs under the same dietary condition. J. Microbiol. Biotechn. 2019, 29, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.K.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2011, 108, 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Hu, J.L.; Nie, Q.X.; Chang, X.; Fang, Q.Y.; Xie, J.H.; Li, H.S.; Nie, S.P. Hypoglycemic mechanism of polysaccharide from Cyclocarya paliurusleaves in type 2 diabetic rats by gut microbiota and host metabolism alteration. Sci. China Life Sci. 2020. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Fernando, S.C.; Purvis, H.T.; Najar, F.Z.; Sukharnikov, L.O.; Krehbiel, C.R.; Nagaraja, T.G.; Roe, B.A.; DeSilva, U. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef]
- Waite, D.W.; Taylor, M.W. Characterizing the avian gut microbiota: Membership, driving influences, and potential function. Front. Microbiol. 2014, 5, 223. [Google Scholar] [CrossRef]
- Nuriel-Ohayon, M.; Neuman, H.; Koren, O. Microbial changes during pregnancy, birth, and infancy. Front. Microbiol. 2016, 7, 1031. [Google Scholar] [CrossRef]
- Shu, Y.L.; Hong, P.; Tang, D.; Qing, H.; Donde, O.O.; Wang, H.; Xiao, B.D.; Wu, H.L. Comparison of intestinal microbes in female and male Chinese concave-eared frogs (Odorrana tormota) and effect of nematode infection on gut bacterial communities. Microbiol. Open 2019, 8, e00749. [Google Scholar] [CrossRef]
- Qin, J.J.; Li, R.Q.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, U59–U70. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, Y.; Wen, Q.; Wang, Y.; Wang, Z.; Tan, Z.; Wu, K. Sex Differences in Intestinal Microbial Composition and Function of Hainan Special Wild Boar. Animals 2020, 10, 1553. https://doi.org/10.3390/ani10091553
Wang X, Zhang Y, Wen Q, Wang Y, Wang Z, Tan Z, Wu K. Sex Differences in Intestinal Microbial Composition and Function of Hainan Special Wild Boar. Animals. 2020; 10(9):1553. https://doi.org/10.3390/ani10091553
Chicago/Turabian StyleWang, Xiaozhe, Ying Zhang, Qiong Wen, Ying Wang, Zhixin Wang, Zhen Tan, and Kebang Wu. 2020. "Sex Differences in Intestinal Microbial Composition and Function of Hainan Special Wild Boar" Animals 10, no. 9: 1553. https://doi.org/10.3390/ani10091553
APA StyleWang, X., Zhang, Y., Wen, Q., Wang, Y., Wang, Z., Tan, Z., & Wu, K. (2020). Sex Differences in Intestinal Microbial Composition and Function of Hainan Special Wild Boar. Animals, 10(9), 1553. https://doi.org/10.3390/ani10091553