Analysis of Whole Genome Resequencing Datasets from a Worldwide Sample of Sheep Breeds to Identify Potential Causal Mutations Influencing Milk Composition Traits

 ,
,  , , and
, , and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
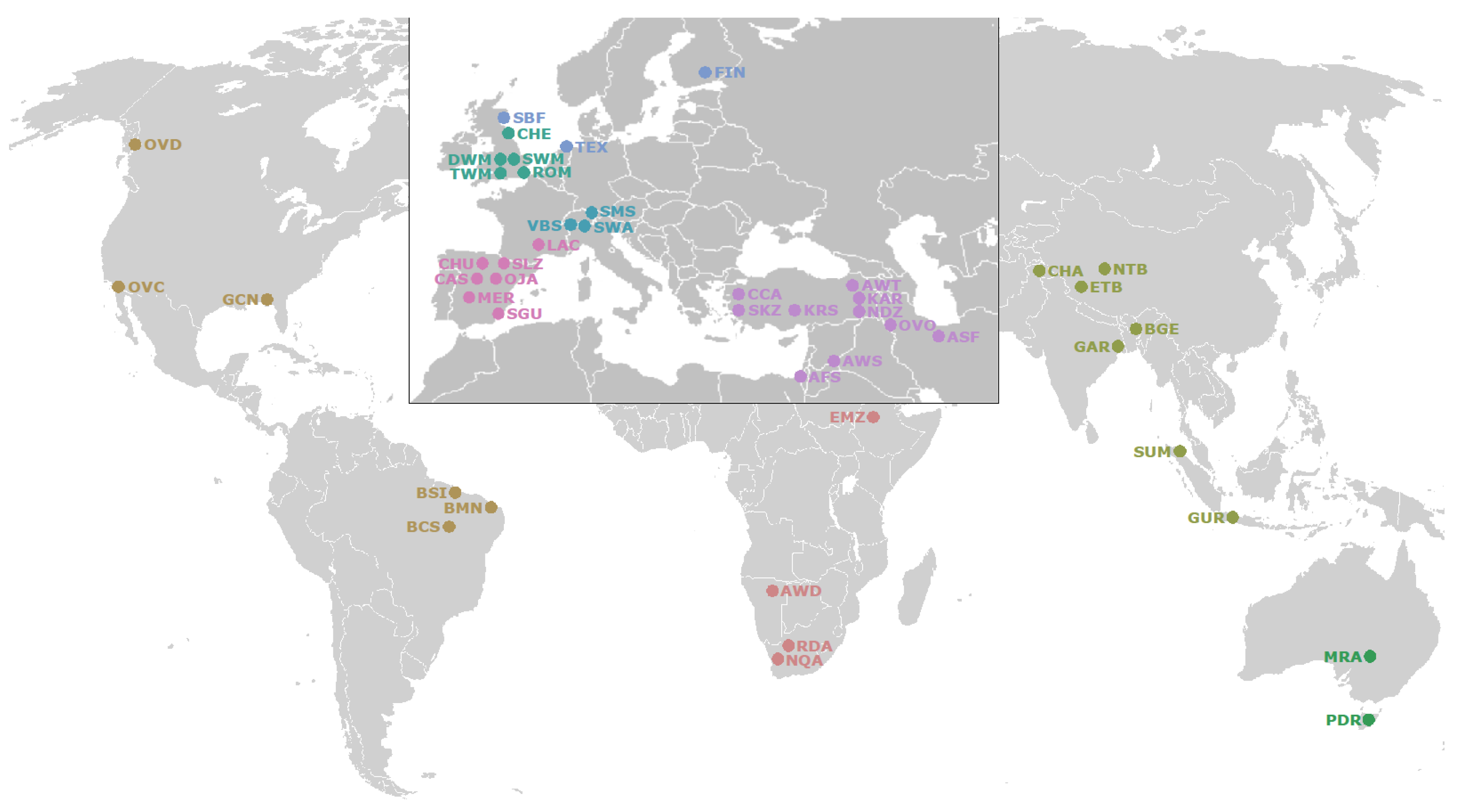
2.1. Whole Genome Resequencing (WGR) Datasets
2.2. Candidate Genes Considered
2.3. WGR Bioinformatics Analysis
2.4. Genetic Diversity and Phylogenetic Analyses
2.5. Functional Annotation and Site Frequency Study for Candidate Gene Variants
3. Results
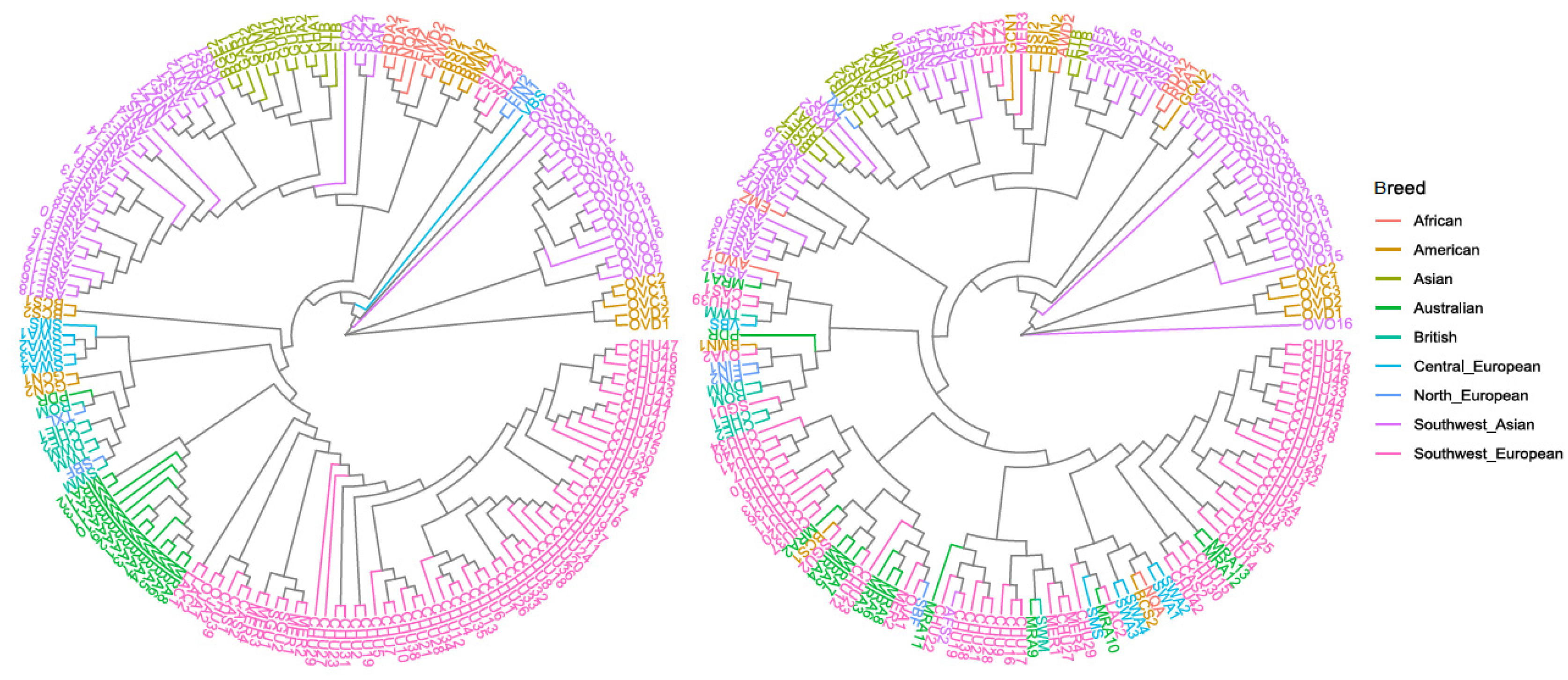
3.1. Genetic Variability and Phylogenetic Relationship between Samples
3.2. Identification of Potential Functionally Relevant Variants in Candidate Genes
4. Discussion
4.1. Variants in Genes Related to Milk Protein Content
4.2. Variants in Genes Involved in Fatty Acid Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chessa, B.; Pereira, F.; Arnaud, F.; Amorim, A.; Goyache, F.; Mainland, I.; Kao, R.R.; Pemberton, J.M.; Beraldi, D.; Stear, M.J.; et al. Revealing the history of sheep domestication using retrovirus integrations. Science 2009, 324, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-Wide Analysis of the World′s Sheep Breeds Reveals High Levels of Historic Mixture and Strong Recent Selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Selvaggi, M.; Laudadio, V.; Dario, C.; Tufarelli, V. Investigating the genetic polymorphism of sheep milk proteins: A useful tool for dairy production. J. Sci. Food Agric. 2014, 94, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Raynal-Ljutovac, K.; Lagriffoul, G.; Paccard, P.; Guillet, I.; Chilliard, Y. Composition of goat and sheep milk products: An update. Small Rumin. Res. 2008, 79, 57–72. [Google Scholar] [CrossRef]
- Sánchez-Mayor, M.; Pong-Wong, R.; Gutiérrez-Gil, B.; Garzón, A.; de la Fuente, L.F.; Arranz, J.J. Phenotypic and genetic parameter estimates of cheese-making traits and their relationships with milk production, composition and functional traits in Spanish Assaf sheep. Livest. Sci. 2019, 228, 76–83. [Google Scholar] [CrossRef]
- Othmane, M.H.; de la Fuente, L.F.; Carriedo, J.A.; San Primitivo, F. Heritability and genetic correlations of test day milk yield and composition, individual laboratory cheese yield, and somatic cell count for dairy ewes. J. Dairy Sci. 2002, 85, 2692–2698. [Google Scholar] [CrossRef]
- Singh, L.V.; Jayakumar, S.; Sharma, A.; Gupta, S.K.; Dixit, S.P.; Gupta, N.; Gupta, S.C. Comparative screening of single nucleotide polymorphisms in β-casein and κ-casein gene in different livestock breeds of India. Meta. Gene. 2015, 4, 85–91. [Google Scholar] [CrossRef]
- Noce, A.; Pazzola, M.; Dettori, M.L.; Amills, M.; Castelló, A.; Cecchinato, A.; Bittante, G.; Vacca, G.M. Variations at regulatory regions of the milk protein genes are associated with milk traits and coagulation properties in the Sarda sheep. Anim. Genet. 2016, 47, 717–726. [Google Scholar] [CrossRef]
- Barillet, F.; Arranz, J.-J.; Carta, A. Mapping quantitative trait loci for milk production and genetic polymorphisms of milk proteins in dairy sheep. Genet. Sel. Evol. 2005, 37 (Suppl. 1), S109–S123. [Google Scholar] [CrossRef]
- Corral, J.M.; Padilla, J.A.; Izquierdo, M. Associations between milk protein genetic polymorphisms and milk production traits in Merino sheep breed. Livest. Sci. 2010, 129, 73–79. [Google Scholar] [CrossRef]
- Giambra, I.J.; Brandt, H.; Erhardt, G. Milk protein variants are highly associated with milk performance traits in East Friesian Dairy and Lacaune sheep. Small Rumin. Res. 2014, 121, 382–394. [Google Scholar] [CrossRef]
- Martin, P.; Szymanowska, M.; Zwierzchowski, L.; Leroux, C.; Martin, P. The impact of genetic polymorphisms on the protein composition of ruminant milks the impact of genetic polymorphisms on the protein composition of ruminant milks. Reprod. Nutr. Dev. 2002, 42, 433. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Azari, M.A.; Zerehdaran, S.; Samiee, R. Effect of β-lactoglobulin and κ-casein genes polymorphism on milk composition in indigenous Zel sheep. Arch. Tierz. 2013, 56, 216–224. [Google Scholar] [CrossRef][Green Version]
- Padilla, P.; Izquierdo, M.; Martínez-Trancón, M.; Parejo, J.C.; Rabasco, A.; Salazar, J.; Padilla, J.Á. Polymorphisms of α-lactoalbumin, β-lactoglobulin and prolactin genes are highly associated with milk composition traits in Spanish Merino sheep. Livest. Sci. 2018, 217, 26–29. [Google Scholar] [CrossRef]
- Garcia-Gámez, E.; Gutiérrez-Gil, B.; Suarez-Vega, A.; de la Fuente, L.F.; Arranz, J.J. Identification of quantitative trait loci underlying milk traits in Spanish dairy sheep using linkage plus combined linkage disequilibrium and linkage analysis approaches. J. Dairy Sci. 2013, 96, 6059–6069. [Google Scholar] [CrossRef]
- García-Gámez, E.; Gutiérrez-Gil, B.; Sahana, G.; Sánchez, J.-P.; Bayón, Y.; Arranz, J.-J.; Jiang, L.; Liu, J.; Sun, D.; Ma, P.; et al. GWA Analysis for Milk Production Traits in Dairy Sheep and Genetic Support for a QTN Influencing Milk Protein Percentage in the LALBA Gene. PLoS ONE 2012, 7, e47782. [Google Scholar]
- Moioli, B.; D’Andrea, M.; Pilla, F. Candidate genes affecting sheep and goat milk quality. Small Rumin. Res. 2007, 68, 179–192. [Google Scholar] [CrossRef]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genom. 2008, 9, 366. [Google Scholar] [CrossRef]
- Crisà, A.; Marchitelli, C.; Pariset, L.; Contarini, G.; Signorelli, F.; Napolitano, F.; Catillo, G.; Valentini, A.; Moioli, B. Exploring polymorphisms and effects of candidate genes on milk fat quality in dairy sheep. J. Dairy Sci. 2010, 93, 3834–3845. [Google Scholar] [CrossRef]
- Moioli, B.; Pilla, F.; Tripaldi, C. Detection of milk protein genetic polymorphisms in order to improve dairy traits in sheep and goats: A review. Small Rumin. Res. 1998, 27, 185–195. [Google Scholar] [CrossRef]
- Suárez-Vega, A.; Gutiérrez-Gil, B.; Arranz, J.J. Transcriptome expression analysis of candidate milk genes affecting cheese-related traits in 2 sheep breeds. J. Dairy Sci. 2016, 99, 6381–6390. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, L.F.; Gabiña, D.; Carolino, N.; Ugarte, E. The awassi and assaf breeds in Spain and Portugal. In Proceedings of the 57th Annual Meeting of the European Association for Animal Production (EAAP), Antalya, Turkey, 17–20 September 2006; p. 79. [Google Scholar]
- De la Fuente, F.; SanPrimitivo, F.; López, T.; Merino, E. Aspectos problemáticos en el programa de selección de la raza churra. Feagas 1996, 10, 46–48. [Google Scholar]
- Ángeles Pérez-Cabal, M.; Legaz, E.; Cervantes, I.; Fernando de la Fuente, L.; Martínez, R.; Goyache, F.; Pablo Gutiérrez, J. Association between body and udder morphological traits and dairy performance in Spanish Assaf sheep Open Access. Arch. Tierz. 2013, 56, 29. [Google Scholar]
- Revilla, I.; Lurueña-Martínez, M.A.; Vivar-Quintana, A.M. Influence of somatic cell counts and breed on physico-chemical and sensory characteristics of hard ewes′-milk cheeses. J. Dairy Res. 2009, 76, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Lurueña-Martínez, M.A.; Palacios, C.; Vivar-Quintana, A.M.; Revilla, I. Effect of the addition of calcium soap to ewes′ diet on fatty acid composition of ewe milk and subcutaneous fat of suckling lambs reared on ewe milk. Meat Sci. 2010, 84, 677–683. [Google Scholar] [CrossRef]
- Sheep genome assembly v3.1. Available online: http://www.ensembl.org/Ovis_aries/Info/Index (accessed on 4 July 2018).
- Andrews, S.; Krueger, F.; Seconds-Pichon, A.; Biggins, F.; Wingett, S. FastQC. A quality control tool for high throughput sequence data. Babraham Bioinformatics. Babraham Inst. 2015, 1, 1. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAM tools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Picard Tools. Available online: http://broadinstitute.github.io/picard/ (accessed on 3 July 2018).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Naval-Sanchez, M.; Nguyen, Q.; McWilliam, S.; Porto-Neto, L.R.; Tellam, R.; Vuocolo, T.; Reverter, A.; Perez-Enciso, M.; Brauning, R.; Clarke, S.; et al. Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds /631/114 /631/181/735 /631/208/177 /631/553 /45/15 /45/23 article. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; De Pristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCF tools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for Inferring Very Large Phylogenies by Using the Neighbor-Joining Method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Gutiérrez-Gil, B.; Esteban-Blanco, C.; Wiener, P.; Chitneedi, P.K.; Suarez-Vega, A.; Arranz, J.-J. High-resolution analysis of selection sweeps identified between fine-wool Merino and coarse-wool Churra sheep breeds. Genet. Sel. Evol. 2017, 49, 81. [Google Scholar] [CrossRef]
- Luigi-Sierra, M.G.; Mármol-Sánchez, E.; Amills, M. Comparing the diversity of the casein genes in the Asian mouflon and domestic sheep. Anim. Genet. 2020, 51, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Juárez, M.; Ramos, M. Physico-chemical characteristics of goat and sheep milk. Small Rumin. Res. 2007, 68, 88–113. [Google Scholar]
- Pazzola, M.; Dettori, M.L.; Pira, E.; Noce, A.; Paschino, P.; Vacca, G.M. Effect of polymorphisms at the casein gene cluster on milk renneting properties of the Sarda goat. Small Rumin. Res. 2014, 117, 124–130. [Google Scholar] [CrossRef]
- Cecchinato, A.; Chessa, S.; Ribeca, C.; Cipolat-Gotet, C.; Bobbo, T.; Casellas, J.; Bittante, G. Genetic variation and effects of candidate-gene polymorphisms on coagulation properties, curd firmness modeling and acidity in milk from Brown Swiss cows. Animal 2015, 9, 1104–1112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suárez-Vega, A.; Gutiérrez-Gil, B.; Klopp, C.; Robert-Granie, C.; Tosser-Klopp, G.; Arranz, J.J. Characterization and comparative analysis of the milk transcriptome in two dairy sheep breeds using RNA sequencing. Sci. Rep. 2015, 5, 18399. [Google Scholar] [CrossRef]
- Hayes, H.C.; Petit, E.J. Mapping of the b-lactoglobulin gene and of an immunoglobulin M heavy chain-like sequence to homoeologous cattle, sheep, and goat chromosomes. Mamm. Genome 1993, 4, 207–210. [Google Scholar] [CrossRef]
- Ramos, A.M.; Matos, C.A.P.; Russo-Almeida, P.A.; Bettencourt, C.M.V.; Matos, J.; Martins, A.; Pinheiro, C.; Rangel-Figueiredo, T. Candidate genes for milk production traits in Portuguese dairy sheep. Small Rumin. Res. 2009, 82, 117–121. [Google Scholar] [CrossRef]
- Selvaggi, M.; Laudadio, V.; Dario, C.; Tufarelli, V. β-Lactoglobulin Gene Polymorphisms in Sheep and Effects on Milk Production Traits: A Review. Adv. Anim. Vet. Sci. 2015, 3, 478–484. [Google Scholar] [CrossRef]
- García-Fernández, M.; Gutiérrez-Gil, B.; García-Gámez, E.; Sánchez, J.P.; Arranz, J.J. The identification of QTL that affect the fatty acid composition of milk on sheep chromosome 11. Anim. Genet. 2010, 41, 324–328. [Google Scholar] [CrossRef]
- Moioli, B.; Scatà, M.C.; de Matteis, G.; Annicchiarico, G.; Catillo, G.; Napolitano, F. The ACACA gene is a potential candidate gene for fat content in sheep milk. Anim. Genet. 2013, 44, 601–603. [Google Scholar] [CrossRef]
- Robenek, H.; Hofnagel, O.; Buers, I.; Lorkowski, S.; Schnoor, M.; Robenek, M.J.; Heid, H.; Troyer, D.; Severs, N.J. Butyrophilin controls milk fat globule secretion. Proc. Natl. Acad. Sci. USA 2006, 103, 10385–10390. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Vega, A.; Gutiérrez-Gil, B.; Klopp, C.; Tosser-Klopp, G.; Arranz, J.J. Variant discovery in the sheep milk transcriptome using RNA sequencing. BMC Genom. 2017, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Hofker, M.H.; Havekes, L.M.; van Dijk, K.W. Living up to a name: The role of the VLDL receptor in lipid metabolism. Curr. Opin. Lipidol. 2001, 12, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Russell, T.D.; Palmer, C.A.; Orlicky, D.J.; Bales, E.S.; Chang, B.H.-J.; Chan, L.; McManaman, J.L. Mammary glands of adipophilin-null mice produce an amino-terminally truncated form of adipophilin that mediates milk lipid droplet formation and secretion. J. Lipid Res. 2008, 49, 206–216. [Google Scholar] [CrossRef]
- Scatà, M.C.; Napolitano, F.; Casu, S.; Carta, A.; de Matteis, G.; Signorelli, F.; Annicchiarico, G.; Catillo, G.; Moioli, B. Ovine acyl CoA: Diacylglycerol acyltransferase 1—molecular characterization, polymorphisms and association with milk traits. Anim. Genet. 2009, 40, 737–742. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Types of Variants According to Different Classification Criteria | Number of Variants within the Candidate Gene Regions | |
|---|---|---|
| Counts SnpEff | Counts VEP | |
| Variants processed | 20,100 | 20,100 |
| SNPs | 17,037 | 16,960 |
| Insertions | 1489 | 1245 |
| Deletions | 1574 | 1396 |
| Indel | 422 | |
| Substitution | 77 | |
| Effects by impact (Only for SNPs) | ||
| High | 5 | 5 |
| Moderate | 203 | 203 |
| Low | 430 | 430 |
| Modifier | 16,378 | 16,378 |
| Effects by functional class (Only for SNPs) | ||
| Missense | 228 | |
| Nonsense | 1 | |
| Silent | 424 | |
| SIFT summary (Only for SNPs) | ||
| Deleterious low confidence | 11 | |
| Tolerated low confidence | 19 | |
| Deleterious | 42 | |
| Tolerated | 150 | |
| Effects by type (Only for SNPs) | ||
| 3 prime UTR | 113 | 113 |
| 5 prime UTR | 59 | 59 |
| Downstream gene | 651 | 651 |
| Frameshift | 0 | 0 |
| Inframe deletion | 0 | 0 |
| Inframe insertion | 0 | 0 |
| Intergenic region | 0 | 0 |
| Intron | 18,579 | 18,574 |
| Missense | 228 | 228 |
| Non coding transcript exon | 12 | 12 |
| Non coding transcript | 137 | 137 |
| Splice acceptor | 4 | 4 |
| Splice donor | 1 | 1 |
| Splice region | 97 | 96 |
| Start lost | 0 | 0 |
| Stop gained | 1 | 1 |
| Stop lost | 0 | 0 |
| Stop retained | 0 | 0 |
| Synonymous | 424 | 424 |
| Upstream gene | 729 | 729 |
| Features | XDH:92215727C>T | LALBA:137390760T>C |
|---|---|---|
| Chromosome | 3 | 3 |
| Position (base pairs) | 92,215,727 | 137,390,760 |
| dbSNP ID | rs429850918 | rs403176291 |
| Reference Allele | C | T |
| Alternate Allele | T | C |
| GeneSymbol | XDH | LALBA |
| Variant | missense | missense |
| BioType | protein coding | protein coding |
| Functional impact (ensemblVEP_Oarv3.1) | MODERATE | MODERATE |
| Functional impact (SIFT_Oarv3.1) | deleterious (0) | deleterious (0.02) |
| Positions in coding sequence | ENSOART00000011926.1:c.1840C>T | ENSOART00000020933.1:c.80T>C |
| Codon change | Cgg/Tgg | gTg/gCg |
| Amino acid substitution | Arg/Trp | Val/Ala |
| Assaf Genotypes (Frequency) | C (0.58), T (0.42) | T (0.92), C (0.08) |
| Churra Genotypes (Frequency) | C (0.97), T (0.03) | T (0.26), C (0.74) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marina, H.; Gutiérrez-Gil, B.; Esteban-Blanco, C.; Suárez-Vega, A.; Pelayo, R.; Arranz, J.J. Analysis of Whole Genome Resequencing Datasets from a Worldwide Sample of Sheep Breeds to Identify Potential Causal Mutations Influencing Milk Composition Traits. Animals 2020, 10, 1542. https://doi.org/10.3390/ani10091542
Marina H, Gutiérrez-Gil B, Esteban-Blanco C, Suárez-Vega A, Pelayo R, Arranz JJ. Analysis of Whole Genome Resequencing Datasets from a Worldwide Sample of Sheep Breeds to Identify Potential Causal Mutations Influencing Milk Composition Traits. Animals. 2020; 10(9):1542. https://doi.org/10.3390/ani10091542
Chicago/Turabian StyleMarina, Héctor, Beatriz Gutiérrez-Gil, Cristina Esteban-Blanco, Aroa Suárez-Vega, Rocío Pelayo, and Juan José Arranz. 2020. "Analysis of Whole Genome Resequencing Datasets from a Worldwide Sample of Sheep Breeds to Identify Potential Causal Mutations Influencing Milk Composition Traits" Animals 10, no. 9: 1542. https://doi.org/10.3390/ani10091542
APA StyleMarina, H., Gutiérrez-Gil, B., Esteban-Blanco, C., Suárez-Vega, A., Pelayo, R., & Arranz, J. J. (2020). Analysis of Whole Genome Resequencing Datasets from a Worldwide Sample of Sheep Breeds to Identify Potential Causal Mutations Influencing Milk Composition Traits. Animals, 10(9), 1542. https://doi.org/10.3390/ani10091542