Ovarian Circular RNAs Associated with High and Low Fertility in Large White Sows during the Follicular and Luteal Phases of the Estrous Cycle

 ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Preparation
2.3. RNA Isolation and Quality Control
2.4. Library Construction and circRNA Sequencing
2.5. Identification of circRNAs and Differential Expression Analysis
2.6. GO and KEGG Pathway Enrichment Analysis
2.7. Protein-Protein Interaction Network Analysis
2.8. Small RNA Sequencing Data Analysis
2.9. Bioinformatic Analysis and Target Prediction
2.10. Reverse-Transcription Real-Time Quantitative PCR
2.11. Statistical Analyses
3. Results
3.1. Characterization of circRNAs in Ovarian Tissues
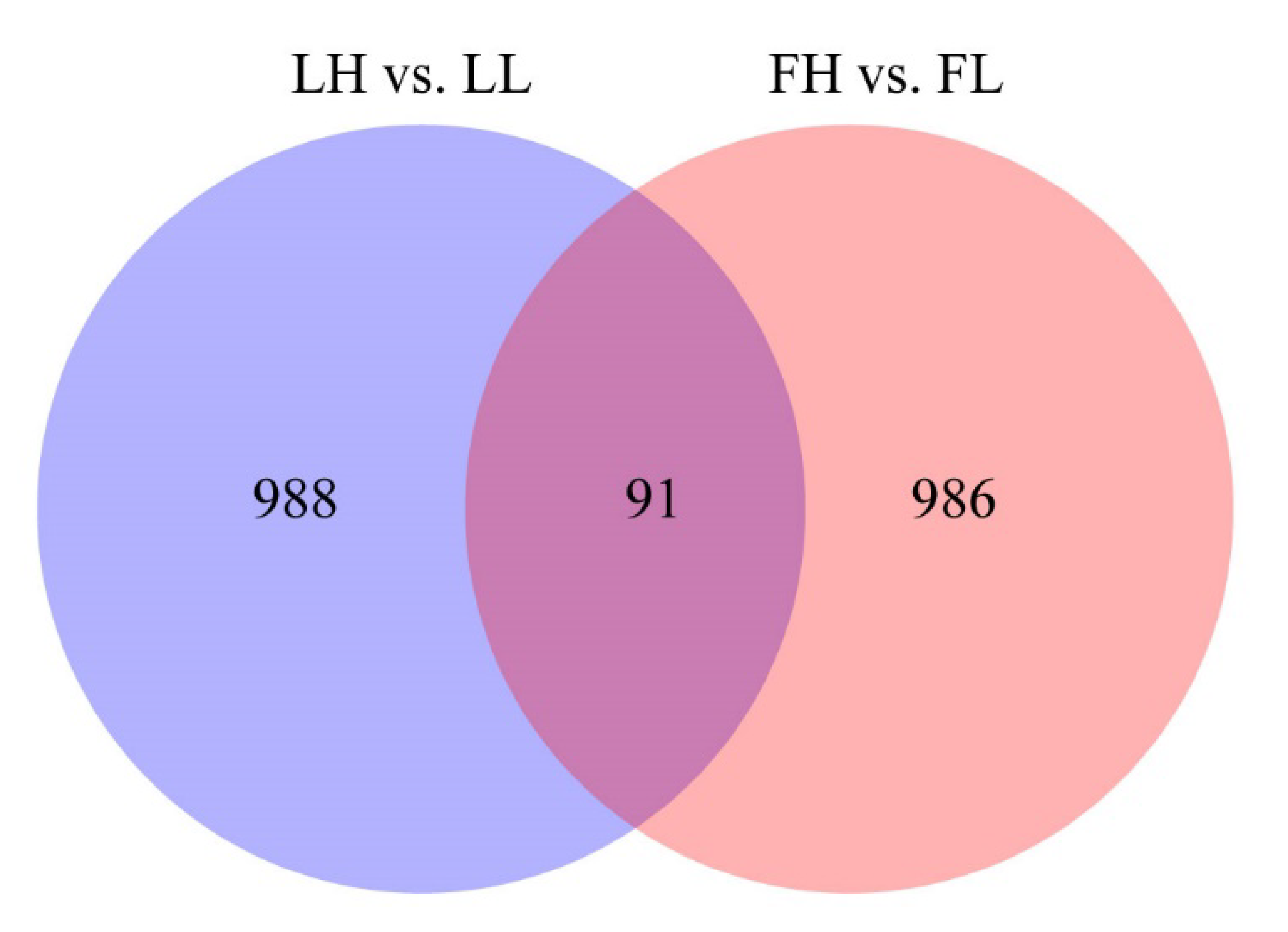
3.2. Differential Expression of circRNAs
3.3. GO and KEGG Enrichment of Host Genes
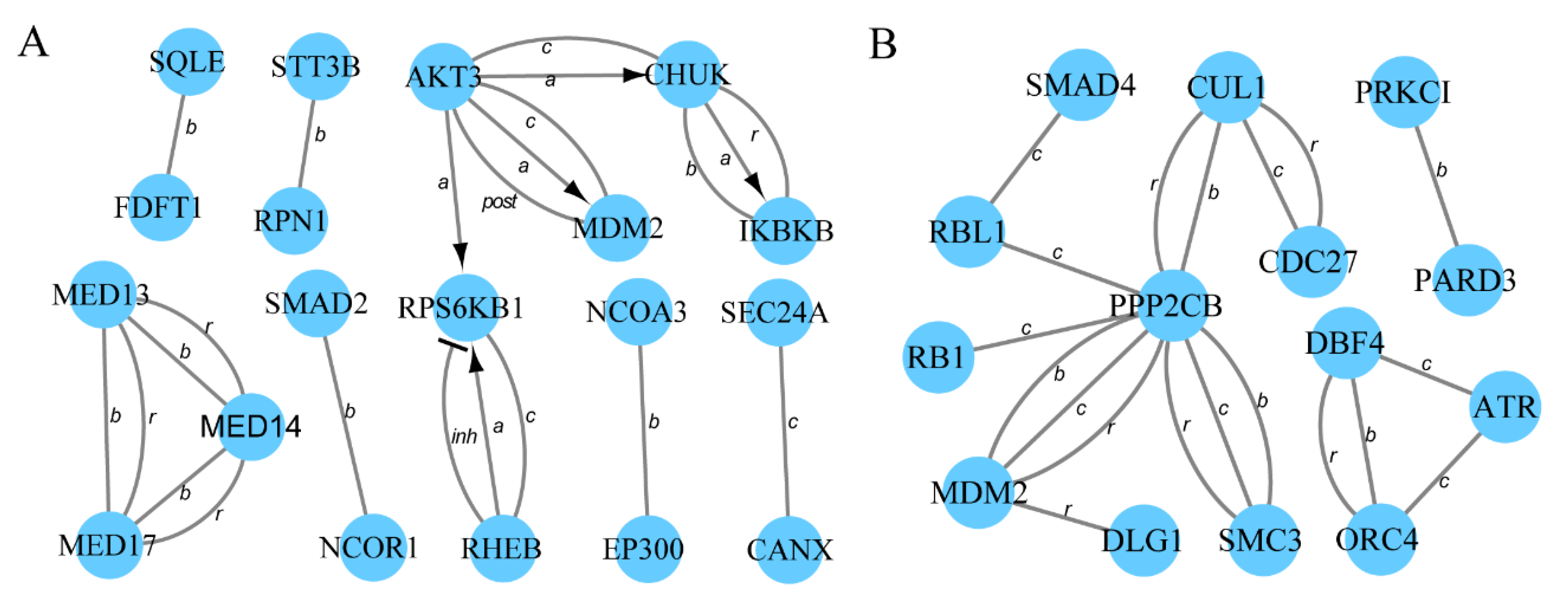
3.4. Protein–Protein Interaction Networks
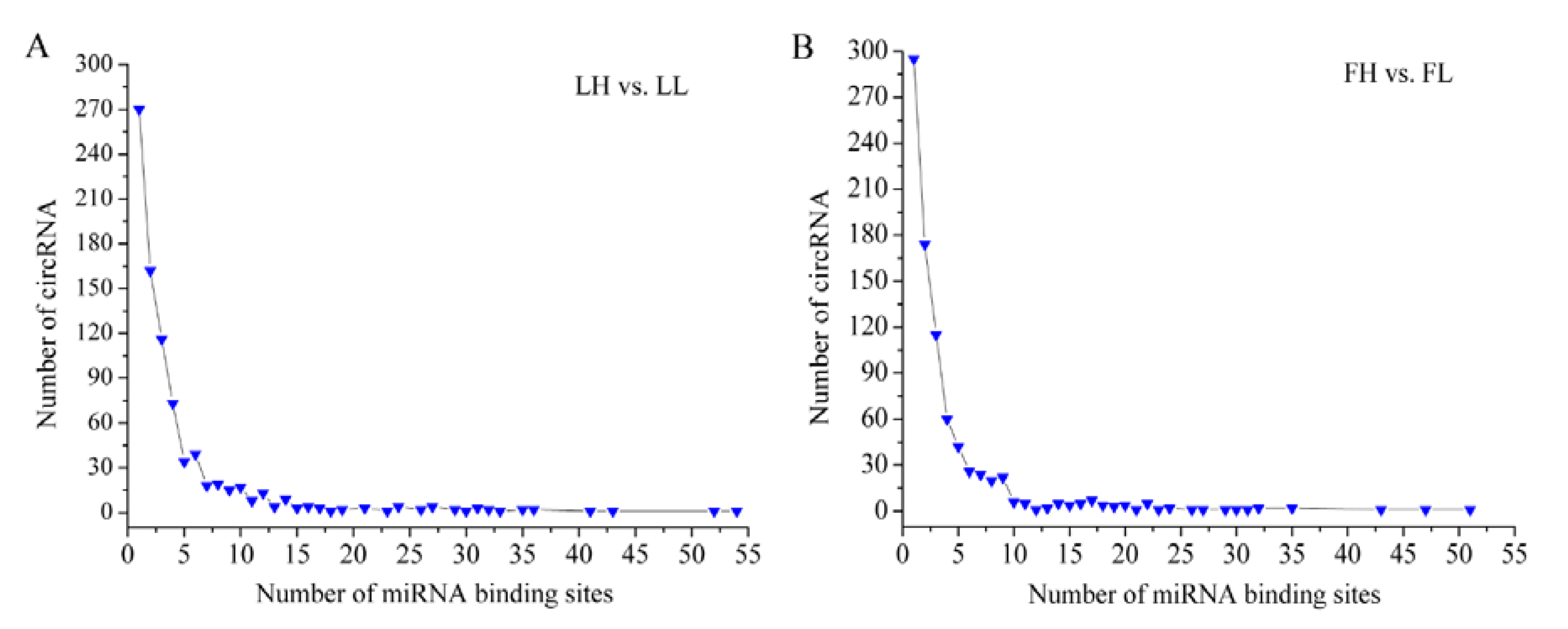
3.5. Prediction of miRNA Binding Sites
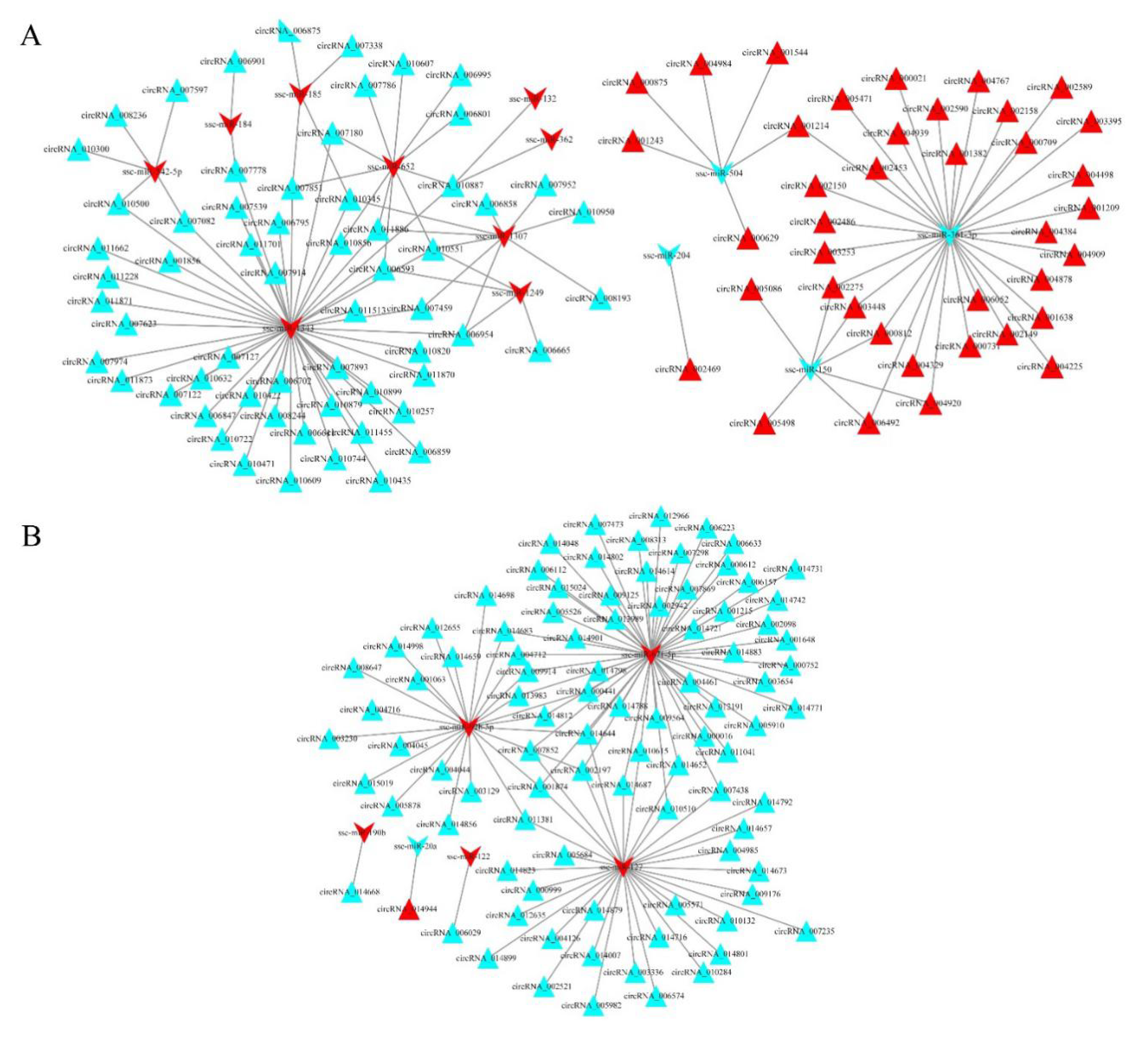
3.6. circRNA–miRNA Interaction Networks
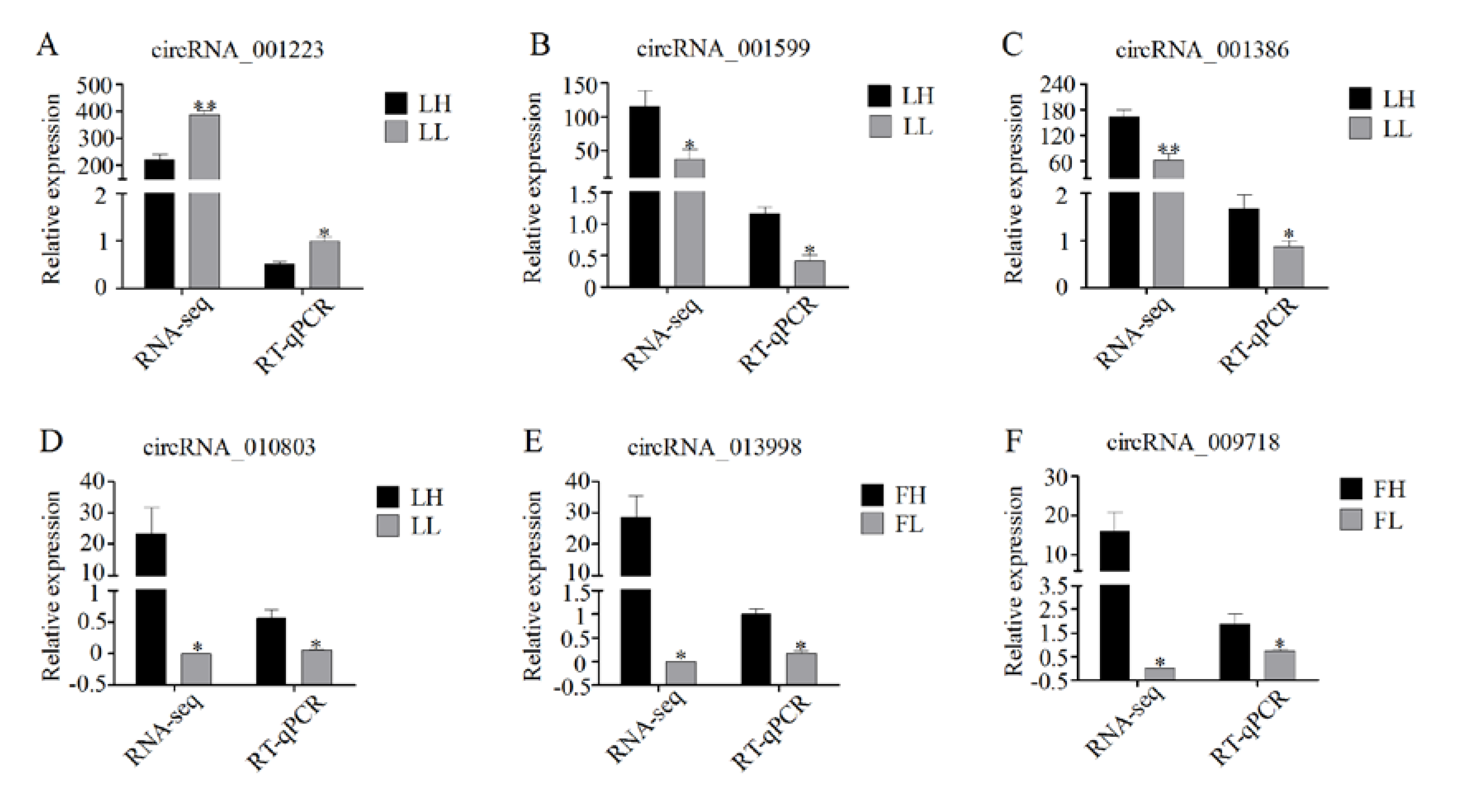
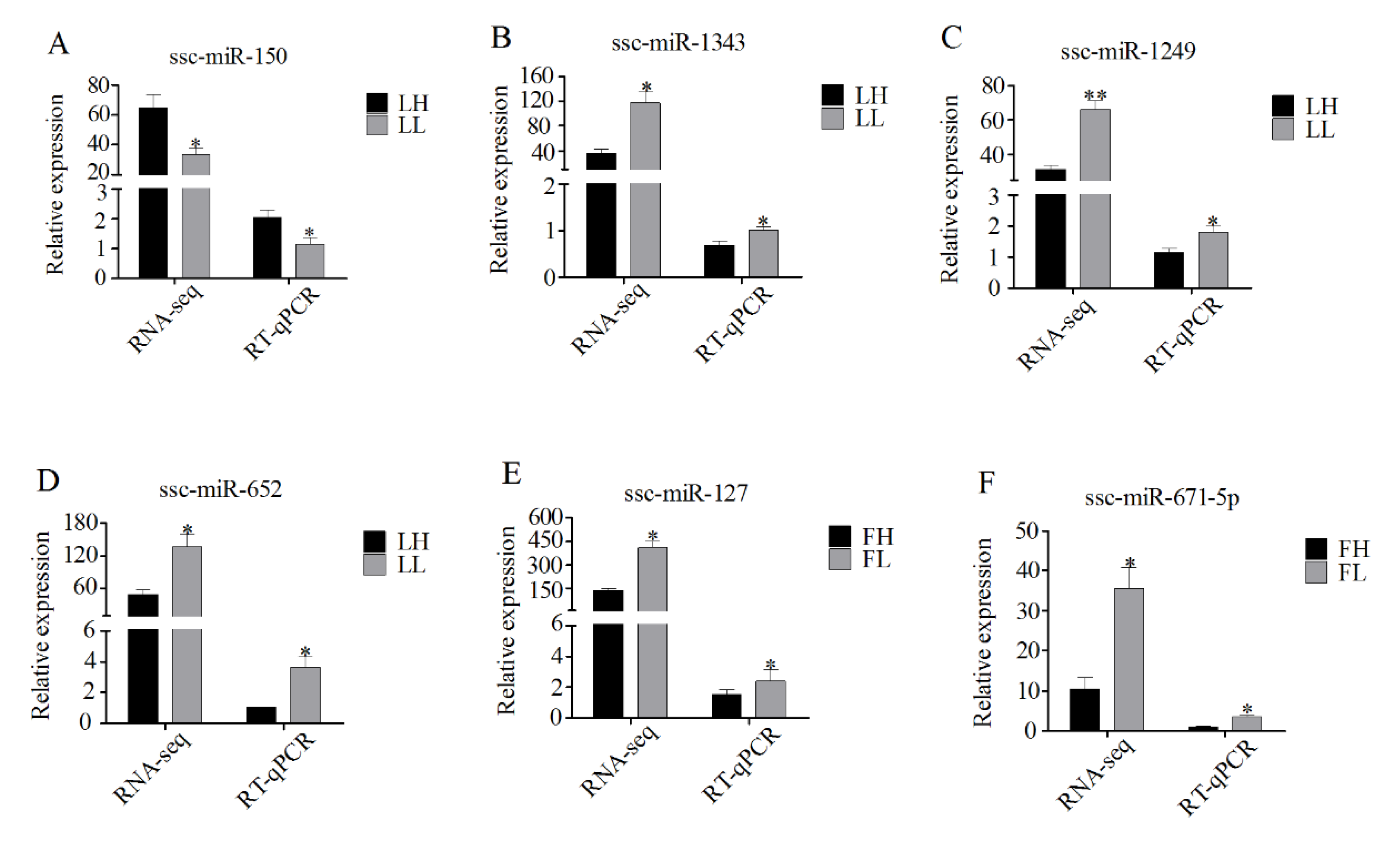
3.7. Validation of RNA-Seq Data Using RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Qu, S.; Yang, X.; Li, X.; Wang, J.; Gao, Y.; Shang, R.; Sun, W.; Dou, K.; Li, H. Circular RNA: A new star of noncoding RNAs. Cancer Lett. 2015, 365, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Ebbesen, K.K.; Kjems, J.; Hansen, T.B. Circular RNAs: Identification, biogenesis and function. Biochim. Biophys. Acta 2016, 1859, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Gruner, H.; Cortés-López, M.; Cooper, D.A.; Bauer, M.; Miura, P. CircRNA accumulation in the aging mouse brain. Sci. Rep. 2016, 6, 38907. [Google Scholar] [CrossRef]
- Cortés-López, M.; Gruner, M.R.; Cooper, D.A.; Gruner, H.N.; Voda, A.; van der Linden, A.M.; Miura, P. Global accumulation of circRNAs during aging in Caenorhabditis elegans. BMC Genom. 2018, 19, 8. [Google Scholar] [CrossRef]
- Liu, Y.C.; Chiu, Y.J.; Li, J.R.; Sun, C.H.; Liu, C.C.; Huang, H.D. Biclustering of transcriptome sequencing data reveals human tissue-specific circular RNAs. BMC Genom. 2018, 19, 958. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Shen, J. Circular RNA participates in the carcinogenesis and the malignant behavior of cancer. RNA Biol. 2017, 14, 514–521. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Liu, J.; Liu, T.; Wang, X.; He, A. Circles reshaping the RNA world: From waste to treasure. Mol. Cancer 2017, 16, 58. [Google Scholar] [CrossRef]
- Yang, J.J.; Tao, H.; Deng, Z.Y.; Lu, C.; Li, J. Non-coding RNA-mediated epigenetic regulation of liver fibrosis. Metabolism 2015, 64, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Xiong, Q.; Zhang, F.; Zhang, N.; Liu, Y.; Suo, X.; Li, X.; Yang, Q.; Chen, M. Circular RNA profiling reveals chi_circ_0008219 function as microRNA sponges in pre-ovulatory ovarian follicles of goats (Capra hircus). Genomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef]
- Chen, I.; Chen, C.Y.; Chuang, T.J. Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdiscip. Rev. RNA 2015, 6, 563–579. [Google Scholar] [CrossRef]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Lai, M.; Li, J.; Zhan, J.; Wen, Q.; Ma, H. Circular RNA expression profiling of granulosa cells in women of reproductive age with polycystic ovary syndrome. Arch. Gynecol. Obstet. 2019, 300, 431–440. [Google Scholar] [CrossRef]
- Cheng, J.; Huang, J.; Yuan, S.; Zhou, S.; Yan, W.; Shen, W.; Chen, Y.; Xia, X.; Luo, A.; Zhu, D.; et al. Circular RNA expression profiling of human granulosa cells during maternal aging reveals novel transcripts associated with assisted reproductive technology outcomes. PLoS ONE 2017, 12, e0177888. [Google Scholar] [CrossRef]
- Chen, X.; Shi, W.; Chen, C. Differential circular RNAs expression in ovary during oviposition in honey bees. Genomics 2019, 111, 598–606. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, L.; Wu, T.; Feng, Y.; Ding, Y.; Ye, P.; Yin, Z. Transcriptomic Analysis of Ovaries from Pigs with High And Low Litter Size. PLoS ONE 2015, 10, e0139514. [Google Scholar] [CrossRef]
- Huang, L.; Yin, Z.J.; Feng, Y.F.; Zhang, X.D.; Wu, T.; Ding, Y.Y.; Ye, P.F.; Fu, K.; Zhang, M.Q. Identification and differential expression of microRNAs in the ovaries of pigs (Sus scrofa) with high and low litter sizes. Anim. Genet. 2016, 47, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Soede, N.M.; Langendijk, P.; Kemp, B. Reproductive cycles in pigs. Anim. Reprod. Sci. 2011, 124, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Mei, S.; Tao, H.; Wang, G.; Su, L.; Jiang, S.; Deng, C.; Xiong, Y.; Li, F. Microarray profiling for differential gene expression in PMSG-hCG stimulated preovulatory ovarian follicles of Chinese Taihu and Large White sows. BMC Genom. 2011, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C (T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ma, Z.; Zhao, H.S.; Zhang, Y.; Liu, X.Y.; Hao, C.F. Novel circular RNA expression in the cumulus cells of patients with polycystic ovary syndrome. Arch. Gynecol. Obstet. 2019, 299, 1715–1725. [Google Scholar] [CrossRef]
- Wang, L.P.; Peng, X.Y.; Lv, X.Q.; Liu, L.; Li, X.L.; He, X.; Lv, F.; Pan, Y.; Wang, L.; Liu, K.F.; et al. High throughput circRNAs sequencing profile of follicle fluid exosomes of polycystic ovary syndrome patients. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Yan, Z.Q.; Jiang, T.T.; Wang, P.F.; Huang, X.Y.; Yang, Q.L.; Sun, W.Y.; Gun, S.B. Circular RNA expression profile of spleen in a Clostridium perfringens type C-induced piglet model of necrotizing enteritis. FEBS Open Bio 2018, 8, 1722–1732. [Google Scholar] [CrossRef]
- Xu, Z.Q.; Yang, M.G.; Liu, H.J.; Su, C.Q. Circular RNA hsa_circ_0003221 (circPTK2) promotes the proliferation and migration of bladder cancer cells. J. Cell Biochem. 2018, 119, 3317–3325. [Google Scholar] [CrossRef]
- Donadeu, F.X.; Schauer, S.N.; Sontakke, S.D. Involvement of miRNAs in ovarian follicular and luteal development. J. Endocrinol. 2012, 215, 323–334. [Google Scholar] [CrossRef]
- Hsueh, A.J.; Kawamura, K.; Cheng, Y.; Fauser, B.C. Intraovarian Control of Early Folliculogenesis. Endocr. Rev. 2015, 36, 1–24. [Google Scholar] [CrossRef]
- Tao, H.; Mei, S.; Sun, X.; Peng, X.; Zhang, X.; Ma, C.; Wang, L.; Hua, L.; Li, F. Associations of TCF12, CTNNAL1 and WNT10B gene polymorphisms with litter size in pigs. Anim. Reprod. Sci. 2013, 140, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Spate, L.D.; Brown, A.N.; Redel, B.K.; Whitworth, K.M.; Murphy, C.N.; Prather, R.S. Dickkopf-Related Protein 1 Inhibits the WNT Signaling Pathway and Improves Pig Oocyte Maturation. PLoS ONE 2014, 9, e95114. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Pangas, S.A.; Jorgez, C.J.; Graff, J.M.; Weinstein, M.; Matzuk, M.M. Redundant roles of SMAD2 and SMAD3 in ovarian granulosa cells in vivo. Mol. Cell Biol. 2008, 28, 7001–7011. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.; Mora, J.M.; Dunlop, C.; Carzaniga, R.; Franks, S.; Fenwick, M.A. Nuclear exclusion of SMAD2/3 in granulosa cells is associated with primordial follicle activation in the mouse ovary. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, J.W.; Prasain, J.K.; Dorand, D.; Yang, Y.; Hoang, H.D.; Vibbert, J.; Kubagawa, H.M.; Miller, M.A. Insulin/FOXO signaling regulates ovarian prostaglandins critical for reproduction. Dev. Cell 2010, 19, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar] [CrossRef]
- Kwintkiewicz, J.; Spaczynski, R.Z.; Foyouzi, N.; Pehlivan, T.; Duleba, A.J. Insulin and oxidative stress modulate proliferation of rat ovarian theca-interstitial cells through diverse signal transduction pathways. Biol. Reprod. 2006, 74, 1034–1040. [Google Scholar] [CrossRef]
- Liu, K. Stem cell factor (SCF)-kit mediated phosphatidylinositol 3 (PI3) kinase signaling during mammalian oocyte growth and early follicular development. Front. Biosci. 2006, 11, 126–135. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, X.; Zhang, M.; Yan, S.; Sun, C.; Xiao, F.; Huang, N.; Yang, X.; Zhao, K.; Zhou, H.; et al. Novel role of FBXW7 circular RNA in repressing glioma tumorigenesis. J. Natl. Cancer Inst. 2018, 110, 304–315. [Google Scholar] [CrossRef]
- Xia, X.; Li, X.; Li, F.; Wu, X. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol. Cancer 2019, 18, 131. [Google Scholar] [CrossRef]
- Portela, V.M.; Dirandeh, E.; Guerrero-Netro, H.M.; Zamberlam, G.; Barreta, M.H.; Goetten, A.F.; Price, C.A. The role of fibroblast growth factor-18 in follicular atresia in cattle. Biol. Reprod. 2015, 92. [Google Scholar] [CrossRef]
- Zielak, A.E.; Canty, M.J.; Forde, N.; Coussens, P.M.; Smith, G.W.; Lonergan, P.; Ireland, J.J.; Evans, A.C.O. Differential expression of genes for transcription factors in theca and granulosa cells following selection of a dominant follicle in cattle. Mol. Reprod. Dev. 2008, 75, 904–914. [Google Scholar] [CrossRef]
- Qu, F.; Wang, F.F.; Yin, R.; Ding, G.L.; El-Prince, M.; Gao, Q.; Shi, B.W.; Pan, H.H.; Huang, Y.T.; Jin, M.; et al. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: Hyperandrogenism induces epigenetic alterations in the granulosa cells. J. Mol. Med. 2012, 90, 911–923. [Google Scholar] [CrossRef]
- Xing, N.; Liang, Y.; Gao, Z.; He, J.; He, X.; Li, H.; Dong, C. Expression and localization of Smad2 and Smad4 proteins in the porcine ovary. Acta Histochem. 2014, 116, 1301–1306. [Google Scholar] [CrossRef]
- Fagerlind, M.; Stålhammar, H.; Olsson, B.; Klinga-Levan, K. Expression of miRNAs in Bull Spermatozoa Correlates with Fertility Rates. Reprod. Domest. Anim. 2015, 50, 587–594. [Google Scholar] [CrossRef]
- McDaneld, T.G.; Kuehn, L.A.; Thomas, M.G.; Snelling, W.M.; Smith, T.P.L.; Pollak, E.J.; Cole, J.B.; Keele, J.W. Genomewide association study of reproductive efficiency in female cattle. J. Anim. Sci. 2014, 92, 1945–1957. [Google Scholar] [CrossRef]
- Ni, W.; You, S.; Cao, Y.; Li, C.; Wei, J.; Wang, D.; Qiao, J.; Zhao, X.; Hu, S.; Quan, R. Aberrant expression of miR-127, miR-21 and miR-16 in placentas of deceased cloned sheep. Res. Vet. Sci. 2016, 105, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Schindler, K.; Schultz, R.M. CDC14A and CDC14B regulate meiotic progression in mouse oocytes. Dev. Biol. 2008, 319, 549. [Google Scholar] [CrossRef]
- Schindler, K.; Schultz, R.M. The CDC14A phosphatase regulates oocyte maturation in mouse. Cell Cycle 2009, 8, 1090–1098. [Google Scholar] [CrossRef]
- Imtiaz, A.; Belyantseva, I.A.; Beirl, A.J.; Fenollar-Ferrer, C.; Bashir, R.; Bukhari, I.; Bouzid, A.; Shaukat, U.; Azaiez, H.; Booth, K.T.; et al. CDC14A phosphatase is essential for hearing and male fertility in mouse and human. Hum. Mol. Genet. 2018, 27, 780–798. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sample ID | Age (Months) | Parities | TNB 3 | p Value | NBA 4 | Estrous Cycle |
|---|---|---|---|---|---|---|---|
| LH 1 | LH1 | 38 | 6 | 17.00 ± 1.45 | 2.65 × 10−5 | 15.83 ± 0.92 | Luteal phase |
| LH2 | 37 | 6 | 16.00 ± 1.51 | 14.67 ± 1.20 | |||
| LH3 | 33 | 5 | 15.50 ± 1.85 | 14.80 ± 0.82 | |||
| LH4 | 33 | 5 | 16.20 ± 2.01 | 14.60 ± 1.02 | |||
| LL 1 | LL1 | 31 | 4 | 9.50 ± 1.84 | 8.75 ± 0.70 | ||
| LL2 | 30 | 4 | 7.33 ± 1.64 | 7.00 ± 0.34 | |||
| LL3 | 23 | 3 | 9.33 ± 1.92 | 8.67 ± 0.43 | |||
| LL4 | 24 | 3 | 9.67 ± 2.10 | 8.67 ± 1.05 | |||
| FH 2 | FH1 | 29 | 4 | 15.75 ± 0.50 | 5.06 × 10−5 | 14.00 ± 0.72 | Follicular phase |
| FH2 | 43 | 7 | 16.71 ± 1.39 | 15.85 ± 1.38 | |||
| FH3 | 42 | 7 | 16.57 ± 1.39 | 14.57 ± 1.05 | |||
| FH4 | 30 | 4 | 17.00 ± 1.84 | 15.75 ± 0.86 | |||
| FL 2 | FL1 | 29 | 4 | 6.00 ± 1.84 | 5.25 ± 0.51 | ||
| FL2 | 23 | 3 | 8.33 ± 2.12 | 7.67 ± 0.40 | |||
| FL3 | 23 | 3 | 4.67 ± 1.53 | 4.33 ± 1.60 | |||
| FL4 | 24 | 3 | 8.33 ± 2.12 | 7.33±0.72 |
| Groups | Gene Name | Primer Sequences (5′-3′) | Amplicon Size (bp) | |
|---|---|---|---|---|
| LH vs. LL | circRNA_001223 | Forward | TTCTGGAGACATCTCGGAGG | 124 |
| Reverse | TCAGGCGGATCTGTTCTTCT | |||
| circRNA_001599 | Forward | GCAGAAATGGCTTCCAATAA | 133 | |
| Reverse | CTTAACCTGGGAAGTGGAACC | |||
| circRNA_001386 | Forward | CACTTGCTTCCGGAGCTTAG | 122 | |
| Reverse | AGCCCTGGGAGTAATTCGA | |||
| circRNA_010803 | Forward | GAATCACACACCACAGGCAC | 143 | |
| Reverse | GCTTTCCAGGCGATCATAAA | |||
| FH vs. FL | circRNA_013998 | Forward | TCCTTCCCAAGAATGTGCTC | 129 |
| Reverse | TGATGGTGCTGAGATCTCCA | |||
| circRNA_009718 | Forward | TCCTGCAATTGAATTCCGTT | 155 | |
| Reverse | TAGGCTCTGGCTTTTTCTCTG | |||
| control | ACTB | Forward | GGCATCCTGACCCTCAAGTA | 100 |
| Reverse | CACGCAGCTCGTTGTAGAAG | |||
| control | U6 | CTCGCTTCGGCAGCACATAT | ||
| LH vs. LL | ssc-miR-150 | TCTCCCAACCCTTGTACCA | ||
| ssc-miR-1343 | ATATCTCCTGGGGCCCGCACTCT | |||
| ssc-miR-1249 | ATATACGCCCTTCCCCCCCTT | |||
| ssc-miR-652 | ATATACAACCCTAGGAGAGGG | |||
| FH vs. FL | ssc-miR-127 | TCGGATCCGTCTGAGCTTGG | ||
| ssc-miR-671-5p | AGGAAGCCCTGGAGGGGCTG | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Xi, J.; Zhou, B.; Zhang, J.; Li, Z.; Liu, Z.; Jia, Q. Ovarian Circular RNAs Associated with High and Low Fertility in Large White Sows during the Follicular and Luteal Phases of the Estrous Cycle. Animals 2020, 10, 696. https://doi.org/10.3390/ani10040696
Hu H, Xi J, Zhou B, Zhang J, Li Z, Liu Z, Jia Q. Ovarian Circular RNAs Associated with High and Low Fertility in Large White Sows during the Follicular and Luteal Phases of the Estrous Cycle. Animals. 2020; 10(4):696. https://doi.org/10.3390/ani10040696
Chicago/Turabian StyleHu, Huiyan, Jianzhong Xi, Bo Zhou, Jing Zhang, Zhiqiang Li, Zhongwu Liu, and Qing Jia. 2020. "Ovarian Circular RNAs Associated with High and Low Fertility in Large White Sows during the Follicular and Luteal Phases of the Estrous Cycle" Animals 10, no. 4: 696. https://doi.org/10.3390/ani10040696
APA StyleHu, H., Xi, J., Zhou, B., Zhang, J., Li, Z., Liu, Z., & Jia, Q. (2020). Ovarian Circular RNAs Associated with High and Low Fertility in Large White Sows during the Follicular and Luteal Phases of the Estrous Cycle. Animals, 10(4), 696. https://doi.org/10.3390/ani10040696