Comprehensive Transcriptional Changes in the Liver of Kanglang White Minnow (Anabarilius grahami) in Response to the Infection of Parasite Ichthyophthirius multifiliis

 and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish and Parasite Resources
2.2. Experimental Design
2.3. RNA Extraction, Library Construction and Transcriptome Sequencing
2.4. Read Alignment and Gene Expression Analysis
2.5. Gene Expression Analysis and Enrichment Analysis
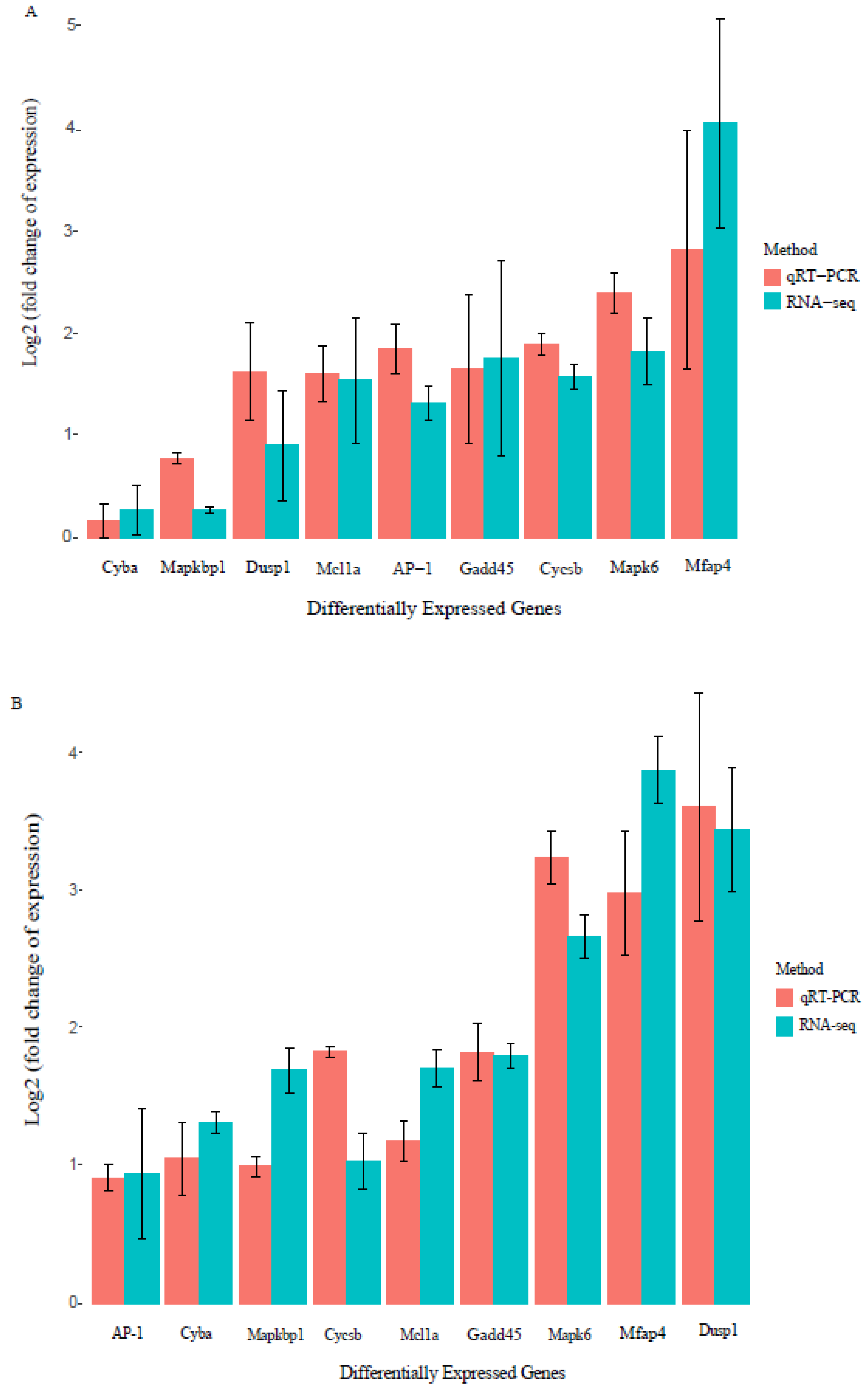
2.6. Validation of Gene Transcription Using Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.7. Identification of Antimicrobial Peptides (AMPs)
3. Results
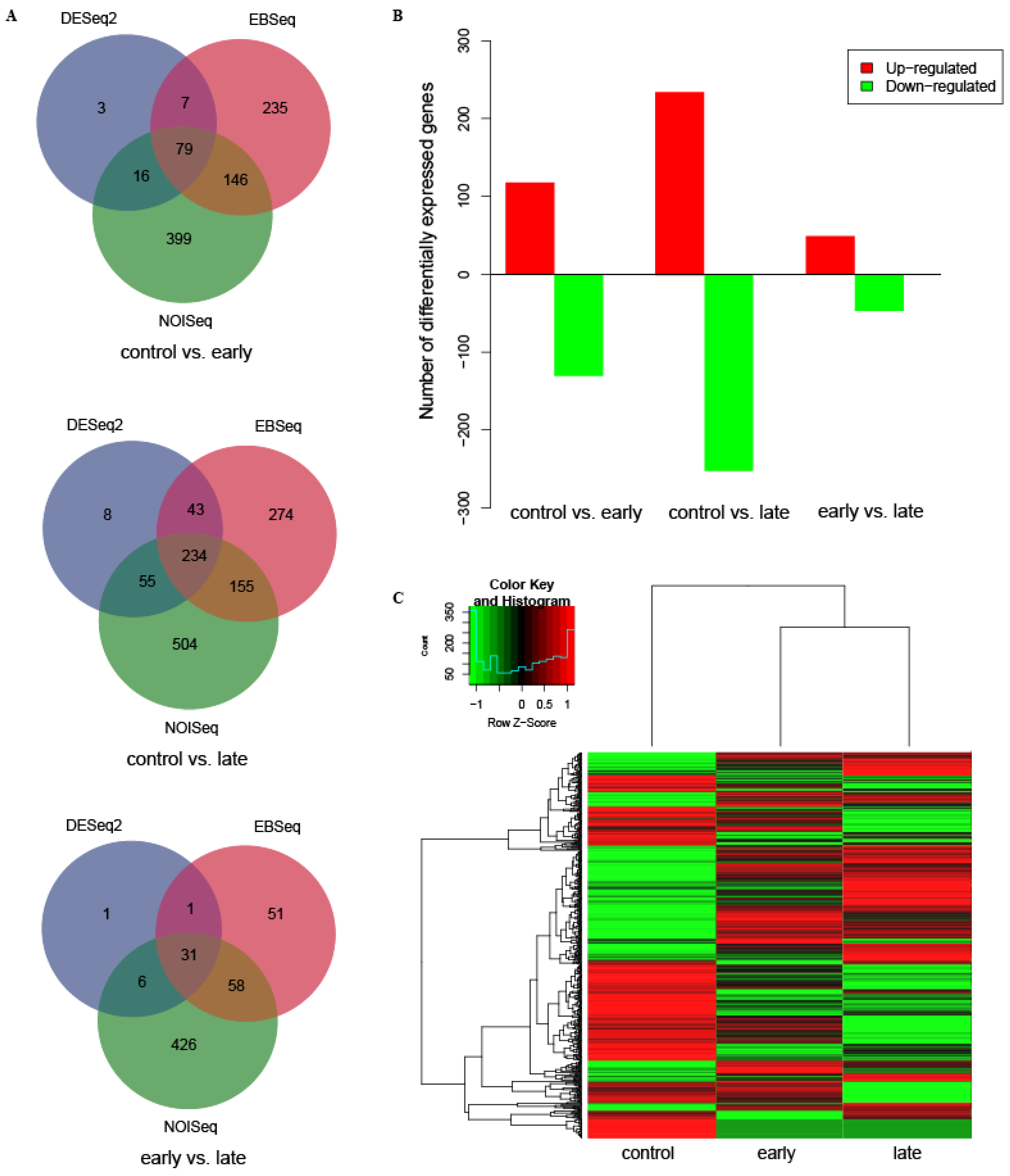
3.1. Identification and Analysis of Differentially Expressed Genes (DEGs)
3.2. Functional Analyses of DEGs Based on GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) Enrichments
3.3. Transcriptional Patterns of the DEGs with Immune Related Functions
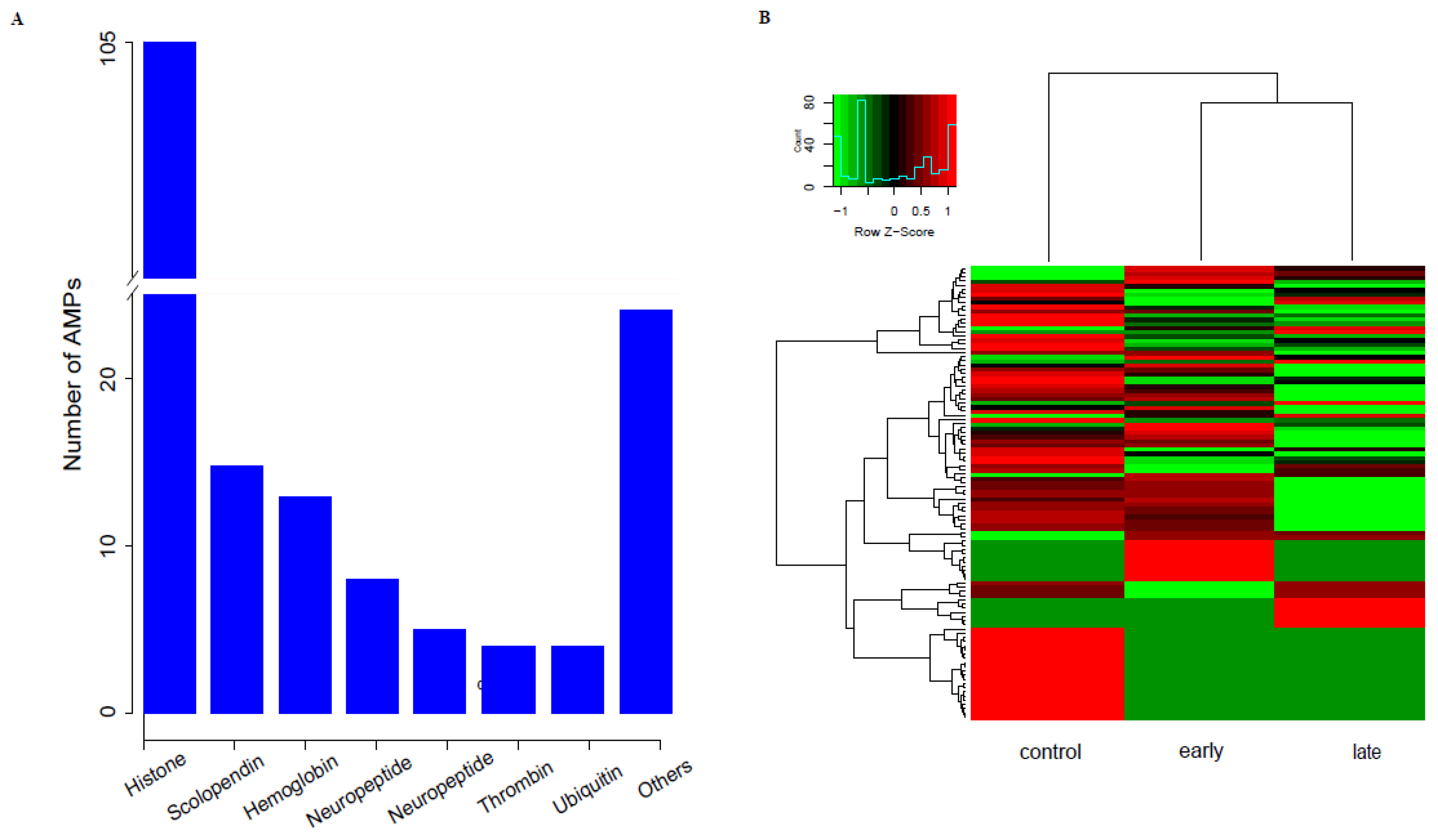
3.4. Identification of AMPs and Their Transcriptional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, W.; Qiu, Y.; Pan, X.; Zhang, Y.; Wang, X.; Lv, Y.; Bian, C.; Li, J.; You, X.; Chen, J.; et al. Genome Assembly for a Yunnan-Guizhou Plateau “3E” Fish, Anabarilius grahami (Regan), and Its Evolutionary and Genetic Applications. Front Genet. 2018, 9, 614. [Google Scholar] [CrossRef]
- Yang, J. Origin and Evolution of some Biological Characters of Anabarilius grahami As Referred To Geological Development of Fuxian Lake. Zool. Res. 1992, 13, 353–360. [Google Scholar]
- Liu, S.; Chen, X.; Yang, J. Threatened fishes of the world: Anabarilius grahami Regan, 1908 (Cyprinidae). Environ. Biol. Fishes 2009, 86, 399–400. [Google Scholar] [CrossRef]
- Qin, J.; Xu, J.; Xie, P. Diet Overlap between the Endemic Fish Anabarilius grahami (Cyprinidae) and the Exotic Noodlefish Neosalanx taihuensis (Salangidae) in Lake Fuxian, China. J. Freshw. Ecol. 2007, 22, 365–370. [Google Scholar] [CrossRef]
- Wang, S. China Species Red List; Higher Education Press: Beijing, China, 2004. [Google Scholar]
- Jiang, Z.; Jiang, J.; Wang, Y.; Zhang, E.; Zhang, Y.; Li, L.; Xie, F.; Cai, B.; Cao, L.; Zheng, G.; et al. Red List of China’s Vertebrates. Biodiv. Sci. 2016, 24, 500–951. [Google Scholar] [CrossRef]
- Jia, Y.; Chen, Y. Effects of Water Trophic Status on Growth Performances of Hemiculter Leucisculus and Anabarilius Grahami. Acta Hydrobiol. Sin. 2008, 32, 333–338. [Google Scholar] [CrossRef]
- Coyne, R.S.; Hannick, L.; Shanmugam, D.; Hostetler, J.B.; Brami, D.; Joardar, V.S.; Johnson, J.; Radune, D.; Singh, I.; Badger, J.H.; et al. Comparative genomics of the pathogenic ciliate Ichthyophthirius multifiliis, its free-living relatives and a host species provide insights into adoption of a parasitic lifestyle and prospects for disease control. Genome Biol. 2011, 12, R100. [Google Scholar] [CrossRef]
- Dickerson, H.W.; Findly, R.C. Immunity to Ichthyophthirius infections in fish: A synopsis. Dev. Comp. Immunol. 2014, 43, 290–299. [Google Scholar] [CrossRef]
- Dickerson, H.; Clark, T. Ichthyophthirius multifiliis: A model of cutaneous infection and immunity in fishes. Immunol. Rev. 1998, 166, 377–384. [Google Scholar] [CrossRef]
- Zaila, K.E.; Cho, D.; Chang, W. Interactions Between Parasitic Ciliates and Their Hosts: Ichthyophthirius multifiliis and Cryptocaryon irritans as Examples. In Biocommunication of Ciliates; Witzany, G., Nowacki, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 327–350. [Google Scholar]
- Alvarez, P.P. Fish immunity and parasite infections: From innate immunity to immunoprophylactic prospects. Vet. Immunol. Immunopathol. 2008, 126, 171–198. [Google Scholar] [CrossRef]
- Mallik, S.; Shahi, N.; Das, P.; Pandey, N.; Haldar, R.S.; Kumar, B.S.; Chandra, S. Occurrence of Ichthyophthirius multifiliis (White spot) infection in snow trout, Schizothorax richardsonii (Gray) and its treatment trial in control condition. Indian J. Anim. Res. 2015, 42, 227–230. [Google Scholar] [CrossRef]
- Louise, V.G.J. Infection and immunity against Ichthyophthirius multifiliis in zebrafish (Danio rerio). Fish Shellfish Immunol. 2016, 57, 335–339. [Google Scholar] [CrossRef]
- Findly, R.C.; Zhao, X.; Noe, J.; Camus, A.C.; Dickerson, H.W. B cell memory following infection and challenge of channel catfish with Ichthyophthirius multifiliis. Dev. Comp. Immunol. 2013, 39, 302–311. [Google Scholar] [CrossRef]
- Zhao, F.; Li, Y.W.; Pan, H.J.; Shi, C.B.; Luo, X.C.; Li, A.X.; Wu, S.Q. Expression profiles of toll-like receptors in channel catfish (Ictalurus punctatus) after infection with Ichthyophthirius multifiliis. Fish Shellfish Immunol. 2013, 35, 993–997. [Google Scholar] [CrossRef]
- Xu, D.H.; Klesius, P.H.; Shoemaker, C.A. Protective immunity of Nile tilapia against Ichthyophthirius multifiliis post-immunization with live theronts and sonicated trophonts. Fish Shellfish Immunol. 2008, 25, 124–127. [Google Scholar] [CrossRef]
- Cross, M.L.; Matthews, R.A. Localized leucocyte response to Ichthyophthirius multifiliis establishment in immune carp Cyprinus carpio L. Vet. Immunol. Immunopathol. 1993, 38, 341–358. [Google Scholar] [CrossRef]
- Gonzalez, S.F.; Buchmann, K.; Nielsen, M.E. Complement expression in common carp (Cyprinus carpio L.) during infection with Ichthyophthirius multifiliis. Dev. Comp. Immunol. 2007, 31, 576–586. [Google Scholar] [CrossRef]
- Gonzalez, S.F.; Buchmann, K.; Nielsen, M.E. Ichthyophthirius multifiliis infection induces massive up-regulation of serum amyloid A in carp (Cyprinus carpio). Vet. Immunol. Immunopathol. 2007, 115, 172–178. [Google Scholar] [CrossRef]
- Gonzalez, S.F.; Buchmann, K.; Nielsen, M.E. Real-time gene expression analysis in carp (Cyprinus carpio L.) skin: Inflammatory responses caused by the ectoparasite Ichthyophthirius multifiliis. Fish Shellfish Immunol. 2007, 22, 641–650. [Google Scholar] [CrossRef]
- Heinecke, R.D.; Buchmann, K. Inflammatory response of rainbow trout Oncorhynchus mykiss (Walbaum, 1792) larvae against Ichthyophthirius multifiliis. Fish Shellfish Immunol. 2013, 34, 521–528. [Google Scholar] [CrossRef]
- Pimenta, A.; Lima, M. Small peptides, big world: Biotechnological potential in neglected bioactive peptides from arthropod venoms. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2005, 11, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yi, Y.; You, X.; Liu, J.; Shi, Q. High-Throughput Identification of Putative Antimicrobial Peptides from Multi-Omics Data of the Lined Seahorse (Hippocampus erectus). Mar. Drugs 2020, 18, 30. [Google Scholar] [CrossRef]
- Yi, Y.; You, X.; Bian, C.; Chen, S.; Lv, Z.; Qiu, L.; Shi, Q. High-Throughput Identification of Antimicrobial Peptides from Amphibious Mudskippers. Mar. Drugs 2017, 15, 364. [Google Scholar] [CrossRef] [PubMed]
- Wung, K.; Chen, X.; Zhang, X.; Li, J.; Yi, Y.; Bian, C.; Shi, Q.; Lin, H.; Shuisheng, L.; Zhang, Y.; et al. Whole Genome Sequencing of the Giant Grouper (Epinephelus lanceolatus) and High-Throughput Screening of Putative Antimicrobial Peptide Genes. Mar. Drugs 2019, 17, 503. [Google Scholar] [CrossRef]
- Han, X.; Wu, X.; Chung, W.; Li, T.; Nekrutenko, A.; Altman, N.S.; Chen, G.; Ma, H. Transcriptome of embryonic and neonatal mouse cortex by high-throughput RNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 12741–12746. [Google Scholar] [CrossRef]
- Li, G.; Zhao, Y.; Liu, Z.; Gao, C.; Yan, F.; Liu, B.; Feng, J. De novo assembly and characterization of the spleen transcriptome of common carp (Cyprinus carpio) using Illumina paired-end sequencing. Fish Shellfish Immunol. 2015, 44, 420–429. [Google Scholar] [CrossRef]
- Wang, P.; Wang, J.; Su, Y.Q.; Mao, Y.; Zhang, J.S.; Wu, C.W.; Ke, Q.Z.; Han, K.H.; Zheng, W.Q.; Xu, N.D. Transcriptome analysis of the Larimichthys crocea liver in response to Cryptocaryon irritans. Fish Shellfish Immunol. 2016, 48, 1–11. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Tarazona, S.; Furio-Tari, P.; Turra, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, 140. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Beissbarth, T.; Speed, T.P. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef]
- Chen, S.; Yang, P.; Jiang, F.; Wei, Y.; Ma, Z.; Kang, L. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS ONE 2010, 5, 15633. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Fu, L.; Shi, J.; Hu, K.; Wang, J.; Wang, W.; Ke, X. Mitogen-activated protein kinase binding protein 1 (MAPKBP1) is an unfavorable prognostic biomarker in cytogenetically normal acute myeloid leukemia. Oncotarget 2015, 6, 8144–8154. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, 1087–1093. [Google Scholar] [CrossRef]
- Gao, Q.; Yue, Y.; Min, M.; Peng, S.; Shi, Z.; Wang, J.; Zhang, T. Time-series transcriptomic analysis of the kelp grouper Epinephelus moara in response to low salinity stress. Anim. Cells Syst. 2018, 22, 234–242. [Google Scholar] [CrossRef]
- Aoki, T.; Takano, T.; Santos, M.; Kondo, H.; Hirono, I. Molecular innate immunity in teleost fish: Review and future perspectives. In Fisheries for Global Welfare and Environment, Memorial Book of the 5th World Fisheries Congress; Terrapub: Tokyo, Japan, 2008; pp. 263–276. [Google Scholar]
- Fukata, M.; Vamadevan, A.S.; Abreu, M.T. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin. Immunol. 2009, 21, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Tong, C.; Feng, C.; Wanghe, K.; Zhao, K. Transcriptomic profiling of Tibetan highland fish (Gymnocypris przewalskii) in response to the infection of parasite ciliate Ichthyophthirius multifiliis. Fish Shellfish Immunol. 2017, 70, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Savan, R.; Sakai, M. Genomics of fish cytokines. Comp. Biochem. Physiol. Part D Genom. Proteom. 2006, 1, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.T.P.; Hokeness, K.L. Cytokine and chemokine networks: Pathways to antiviral defense. Curr. Top Microbiol. Immunol. 2006, 303, 29–46. [Google Scholar] [CrossRef]
- Song, X.; He, X.; Li, X.; Qian, Y. The roles and functional mechanisms of interleukin-17 family cytokines in mucosal immunity. Cell. Mol. Immunol. 2016, 13, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, 000034. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Xu, D.; Moreira, G.; Shoemaker, C.; Zhang, D.; Beck, B. Expression of immune genes in systemic and mucosal immune tissues of channel catfish vaccinated with live theronts of Ichthyophthirius multifiliis. Fish Shellfish Immunol. 2017, 66, 540–547. [Google Scholar] [CrossRef]
- Roelands, J.; Garand, M.; Hinchcliff, E.; Ma, Y.; Shah, P.; Toufiq, M.; Alfaki, M.; Hendrickx, W.; Boughorbel, S.; Rinchai, D.; et al. Long-Chain Acyl-CoA Synthetase 1 Role in Sepsis and Immunity: Perspectives From a Parallel Review of Public Transcriptome Datasets and of the Literature. Front Immunol. 2019, 10, 2410. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Bhat, A.; Gomis, S.; Potter, A.; Tikoo, S. Role of Hsp90 in CpG ODN mediated immunostimulation in avian macrophages. Mol. Immunol. 2010, 47, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Bustillo, M.E.; Fischer, A.L.; LaBouyer, M.A.; Klaips, J.A.; Webb, A.C.; Elmore, D.E. Modular analysis of hipposin, a histone-derived antimicrobial peptide consisting of membrane translocating and membrane permeabilizing fragments. Biochim. Biophys. Acta 2014, 1838, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.; van Eijk, M.; Haagsman, H.P.; Hartshorn, K.L. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol. 2016, 11, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Dai, J.; Zay, K.; Karsan, A.; Hendricks, R.; Yee, C.; Martin, R.; MacKenzie, R.; Xie, C.; Zhang, L.; et al. Alpha-1-Antitrypsin and a Broad Spectrum Metalloprotease Inhibitor, RS113456, Have Similar Acute Anti-Inflammatory Effects. Lab. Investig. J. Tech. Methods Pathol. 2001, 81, 1119–1131. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pairwise Comparison | DEGs | GO Terms | KEGG Pathway | |||||
|---|---|---|---|---|---|---|---|---|
| Up-Regulated | Down-Regulated | Total | Biological Process | Cellular Component | Molecular Function | Total | ||
| control vs. early-infected | 118 | 130 | 248 | 20 | 1 | 11 | 32 | 30 |
| control vs. late-infected | 235 | 252 | 487 | 54 | 1 | 23 | 78 | 28 |
| early vs. late-infected | 49 | 47 | 96 | 11 | 1 | 19 | 31 | 18 |
| Total | 402 | 429 | 831 | 85 | 3 | 53 | 128 * | 71 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.; Yin, Y.; Ruan, Z.; Gao, Y.; Bian, C.; Chen, J.; Wang, X.; Pan, X.; Yang, J.; Shi, Q.; et al. Comprehensive Transcriptional Changes in the Liver of Kanglang White Minnow (Anabarilius grahami) in Response to the Infection of Parasite Ichthyophthirius multifiliis. Animals 2020, 10, 681. https://doi.org/10.3390/ani10040681
Qiu Y, Yin Y, Ruan Z, Gao Y, Bian C, Chen J, Wang X, Pan X, Yang J, Shi Q, et al. Comprehensive Transcriptional Changes in the Liver of Kanglang White Minnow (Anabarilius grahami) in Response to the Infection of Parasite Ichthyophthirius multifiliis. Animals. 2020; 10(4):681. https://doi.org/10.3390/ani10040681
Chicago/Turabian StyleQiu, Ying, Yanhui Yin, Zhiqiang Ruan, Yu Gao, Chao Bian, Jieming Chen, Xiaoai Wang, Xiaofu Pan, Junxing Yang, Qiong Shi, and et al. 2020. "Comprehensive Transcriptional Changes in the Liver of Kanglang White Minnow (Anabarilius grahami) in Response to the Infection of Parasite Ichthyophthirius multifiliis" Animals 10, no. 4: 681. https://doi.org/10.3390/ani10040681
APA StyleQiu, Y., Yin, Y., Ruan, Z., Gao, Y., Bian, C., Chen, J., Wang, X., Pan, X., Yang, J., Shi, Q., & Jiang, W. (2020). Comprehensive Transcriptional Changes in the Liver of Kanglang White Minnow (Anabarilius grahami) in Response to the Infection of Parasite Ichthyophthirius multifiliis. Animals, 10(4), 681. https://doi.org/10.3390/ani10040681