Effects of High Forage/Concentrate Diet on Volatile Fatty Acid Production and the Microorganisms Involved in VFA Production in Cow Rumen

Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Design, Animals Feeding, and Sample Collection
2.3. Sample Measurements
2.4. RNA Extraction, RNA Reverses Transcribed, qPCR Primer Design and Analysis
2.5. DNA Extraction, Metagenome Library Preparation and Sequencing
2.6. Metagenome Assembly, Taxonomic and KEGG Analysis
2.7. Statistical Analysis
3. Results
3.1. Rumen Fermentation Parameters of Cows Feed Two Different Diets
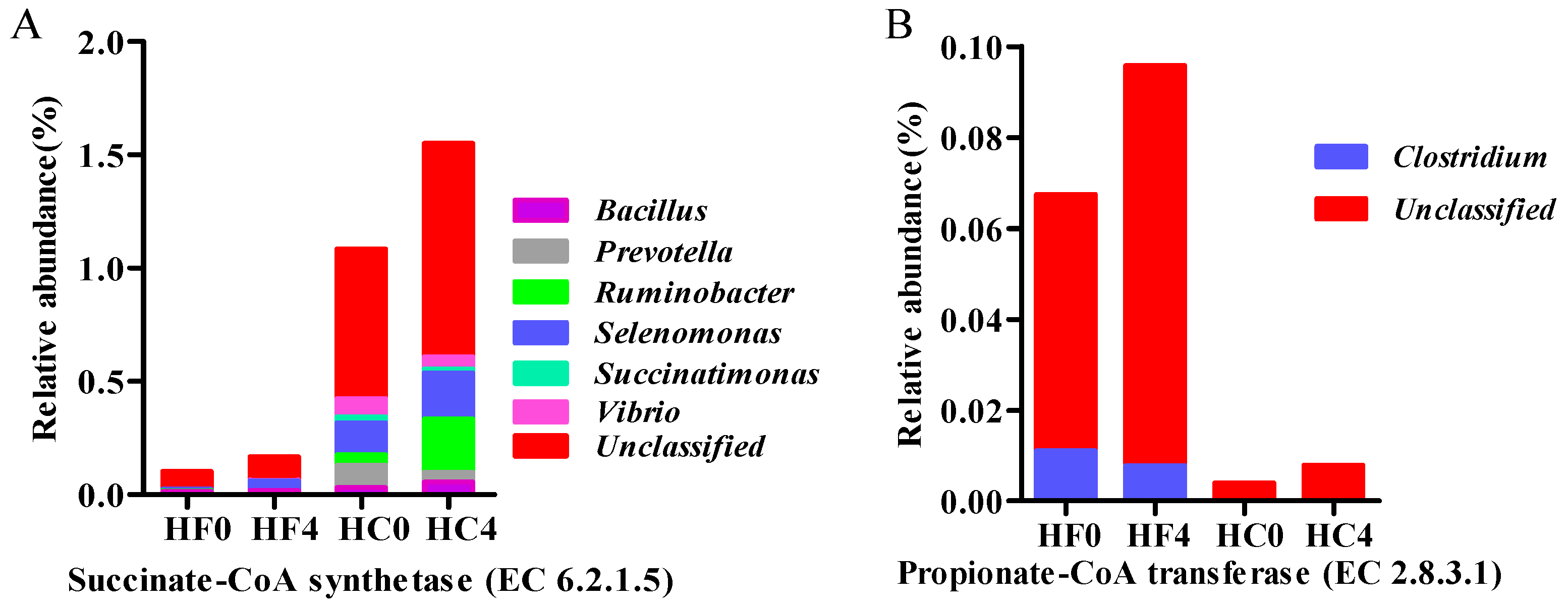
3.2. Genes Directly Involved in Propionate
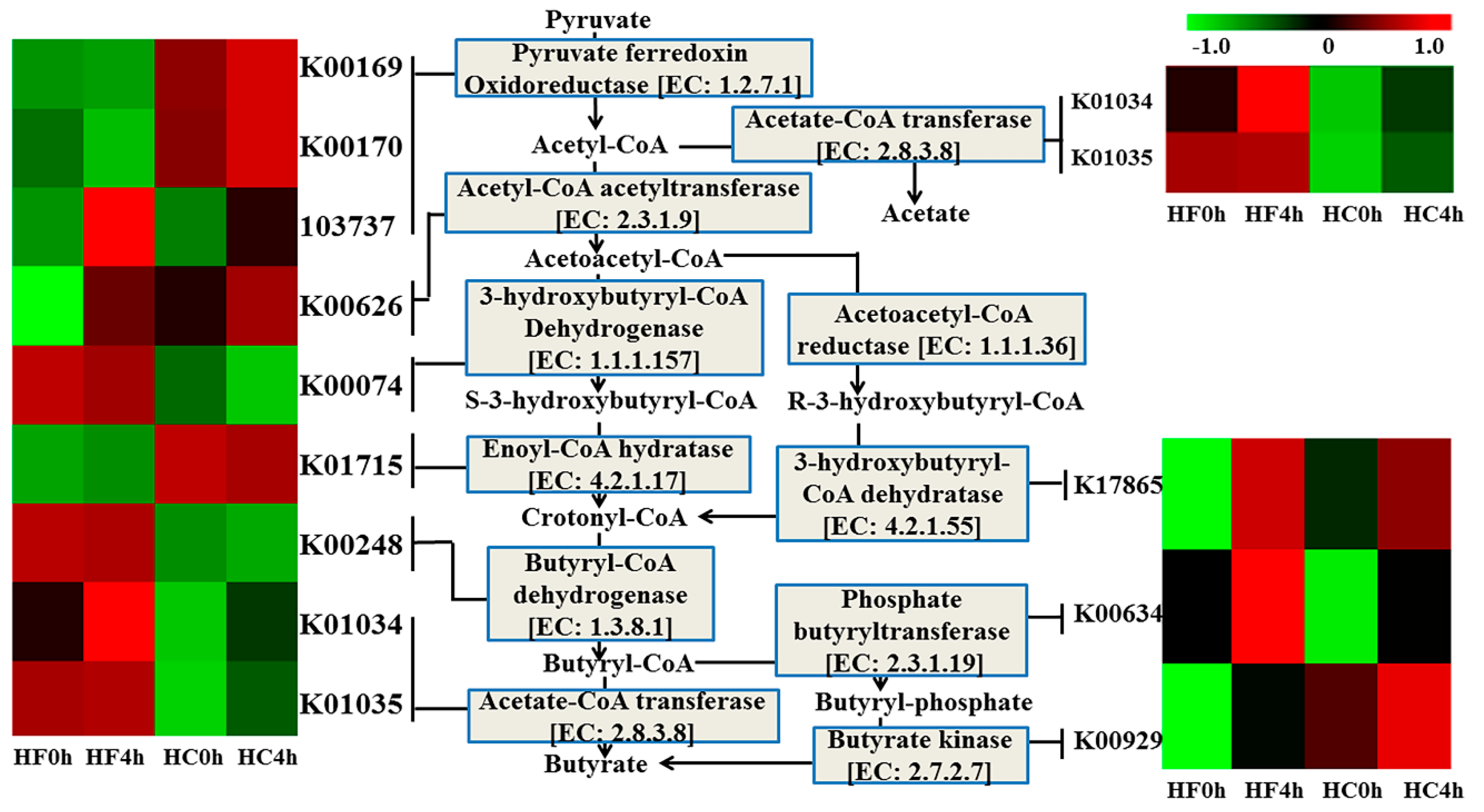
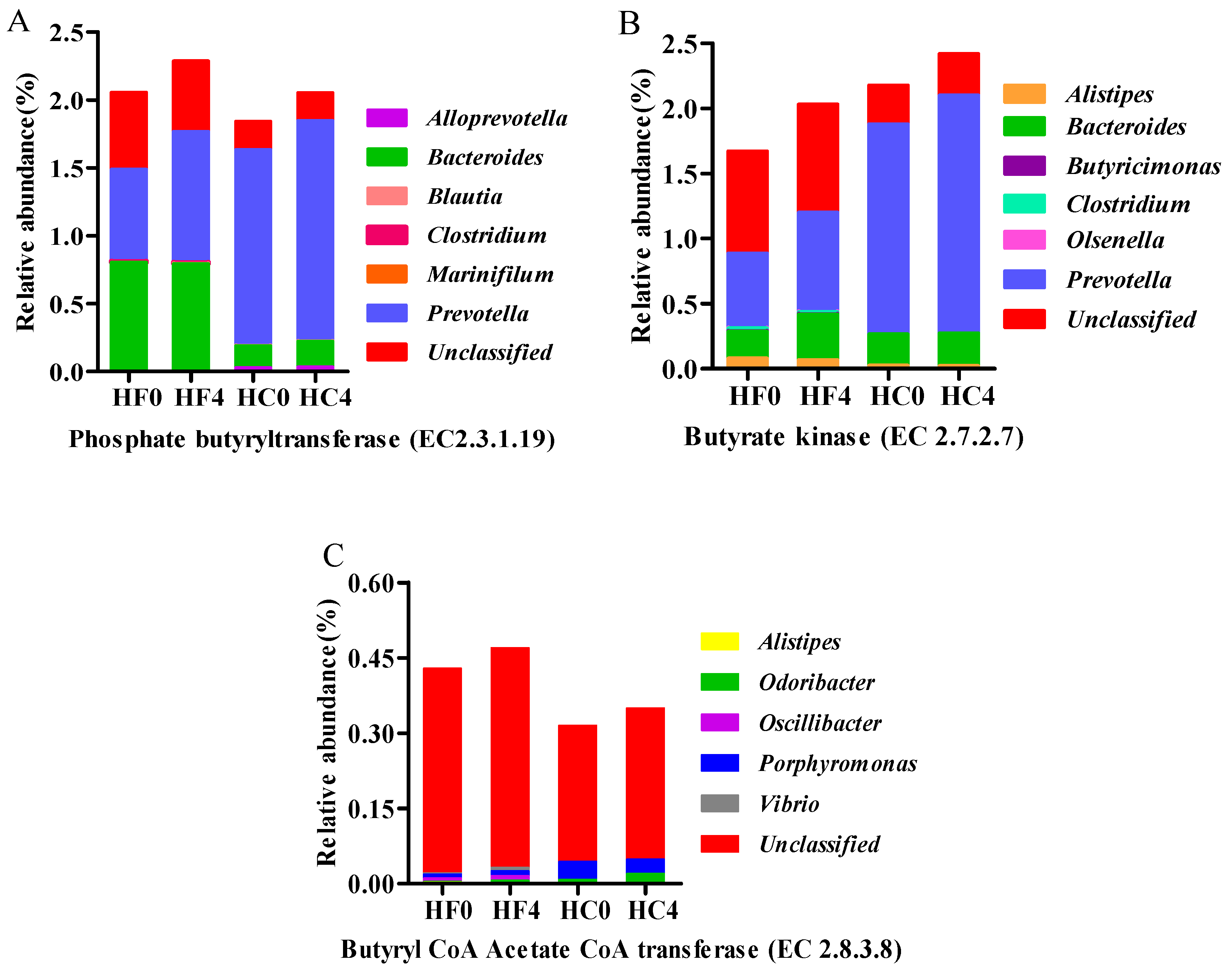
3.3. Genes Directly Involved in Butyrate
3.4. The Major Microbial Material Involved in Cellulose and Volatile Fatty Acid Metabolism Gene Characterization and Quantification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Russell, J.B.; O’Connor, J.D.; Fox, D.G.; Van Soest, P.J.; Sniffen, C.J. A net carbohydrate and protein system for evaluating cattle diets: I. ruminal fermentation. J. Anim. Sci. 1992, 70, 3551–3561. [Google Scholar] [CrossRef] [PubMed]
- Ceconi, I.; Ruiz-Moreno, M.; DiLorenzo, N.; DiCostanzo, A.; Crawford, G.I. Effect of urea inclusion in diets containing corn dried distillers grains on feedlot cattle performance, carcass characteristics, ruminal fermentation, total tract digestibility, and purine derivatives-to-creatinine index. J. Anim Sci. 2015, 93, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.D. The fermentation of soluble carbohydrates in rumen contents of cows fed diets containing a large proportion of hay. Br. J. Nutr. 1968, 22, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, X.; Zhao, C.; Hu, P.; Chen, H.; Liu, Z.; Liu, G.; Wang, Z. Correlation between composition of the bacterial community and concentration of volatile fatty acids in the rumen during the transition period and ketosis in dairy cows. Appl. Environ. Microbiol. 2012, 78, 2386–2392. [Google Scholar] [CrossRef]
- Alstrup, L.; Søegaard, K.; Weisbjerg, M.R. Effects of maturity and harvest season of grass-clover silage and of forage-to-concentrate ratio on milk production of dairy cows. J. Dairy Sci. 2016, 99, 328–340. [Google Scholar] [CrossRef]
- Bharanidharan, R.; Arokiyaraj, S.; Kim, E.B.; Lee, C.H.; Woo, Y.W.; Na, Y.; Kim, K.H. Ruminal methane emissions, metabolic, and microbial profile of Holstein steers fed forage and concentrate, separately or as a total mixed ration. PloS ONE 2018, 13, e0202446. [Google Scholar] [CrossRef]
- Welkie, D.G.; Stevenson, D.M.; Weimer, P.J. ARISA analysis of ruminal bacterial community dynamics in lactating dairy cows during the feeding cycle. Anaerobe. 2010, 16, 94–100. [Google Scholar] [CrossRef]
- Ribeiro, R.C.; Villela, S.D.J.; Valadares Filho, S.C.; Santos, S.A.; Ribeiro, K.G.; Detmann, E.; Martins, P.G.M.A. Effects of roughage sources produced in a tropical environment on forage intake, and ruminal and microbial parameters. J. Anim Sci. 2015, 93, 2363–2374. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, H.; Wang, Y.; Li, S.; Cao, Z.; Ji, S.; Zhang, H. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in Holstein heifers. Front. Microbiol. 2017, 8, 2206–2214. [Google Scholar] [CrossRef]
- Penner, G.B.; Taniguchi, M.; Guan, L.L.; Beauchemin, K.A.; Oba, M. Effect of dietary forage to concentrate ratio on volatile fatty acid absorption and the expression of genes related to volatile fatty acid absorption and metabolism in ruminal tissue. J. Dairy Sci. 2009, 92, 2767–2781. [Google Scholar] [CrossRef]
- Kljak, K.; Pino, F.; Heinrichs, A.J. Effect of forage to concentrate ratio with sorghum silage as a source of forage on rumen fermentation, N balance, and purine derivative excretion in limit-fed dairy heifers. J. Dairy Sci. 2017, 100, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.R.; Ventto, L.; Kairenius, P.; Stefański, T.; Leskinen, H.; Tapio, I.; Shingfield, K.J. Dietary forage to concentrate ratio and sunflower oil supplement alter rumen fermentation, ruminal methane emissions, and nutrient utilization in lactating cows. Translat Anim Sci. 2017, 1, 277–286. [Google Scholar] [CrossRef]
- Mao, S.Y.; Huo, W.J.; Zhu, W.Y. Microbiome–metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ. Microbiol. 2016, 18, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Zhao, S.; Wang, P.; Zheng, N.; Bu, D.; Beckers, Y.; Wang, J. Insights into abundant rumen ureolytic bacterial community using rumen simulation system. Front. Microbiol. 2016, 7, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.J.; Li, S.L.; Xing, J.J.; Ma, M.; Wang, L.L. Effects of maize grain and lucerne particle size on ruminal fermentation, digestibility and performance of cows in midlactation. J. Anim. Physiol. Anim. Nutr. 2008, 92, 157. [Google Scholar] [CrossRef]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.; Mcsweeney, C.S. An efficient RNA extraction method for estimating gut microbial diversity by polymerase chain reaction. Curr. Microbiol. 2009, 58, 464–471. [Google Scholar] [CrossRef]
- Khafipour, E.; Li, S.; Plaizier, J.C.; Krause, D.O. Rumen microbiome composition determined using two nutritional models of subacute ruminal acidosis. Appl Environ. Microbiol. 2009, 75, 7115–7124. [Google Scholar] [CrossRef]
- Edwards, J.E.; Kingston-Smith, A.H.; Jimenez, H.R.; Huws, S.A.; Skøt, K.P.; Griffith, G.W.; Theodorou, M.K. Dynamics of initial colonization of nonconserved perennial ryegrass by anaerobic fungi in the bovine rumen. FEMS Microbiol. Ecol. 2010, 66, 537–545. [Google Scholar] [CrossRef]
- Guo, J.X.; Lu, Y.; Zhu, W.Y.; Denman, S.E.; Mcsweeney, C.S. Effect of tea saponin on methanogenesis, microbial community structure and expression of mcrA gene, in cultures of rumen micro-organisms. Lett. Appl. Microbiol. 2010, 47, 421–426. [Google Scholar] [CrossRef]
- Doyle, J.J.; Dickson, E.E. Preservation of plant samples for DNA restriction endonuclease analysis. Taxon 1987, 36, 715–722. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; Tang, J. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18–24. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Yue, Z.; He, W.; Yang, X.; Yang, G.; Xie, M.; Douglas, C.J. A heterozygous moth genome provides insights into herbivory and detoxification. Nat. Genet. 2013, 45, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lomsadze, A.; Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef]
- Thurston, B.; Dawson, K.A.; Strobel, H.J. Cellobiose versus glucose utilization by the ruminal bacterium Ruminococcus albus. Appl. Environ. Microbiol. 1993, 59, 2631–2637. [Google Scholar] [CrossRef]
- Hristov, A.N.; Zaman, S.; Schneider, C. Effect of dietary concentrate on rumen fermentation, digestibility, and nitrogen losses in dairy cows. J. Dairy Sci. 2010, 93, 4211–4222. [Google Scholar]
- Ash, R.W.; Dobson, A. The effect of absorption on the acidity of rumen contents. J. Physiol. 1963, 169, 39–61. [Google Scholar] [CrossRef]
- Blaxter, K.L. The Energy Metabolism of Ruminants. Digestive Physiology and Metabolism in Ruminants: Proceedings of the 5th; Springer: Springfield, UK, 1962. [Google Scholar]
- Wang, C.; Liu, Q.; Guo, G.; Huo, W.J.; Ma, L.; Zhang, Y.L.; Wang, H. Effects of rumen-protected folic acid on ruminal fermentation, microbial enzyme activity, cellulolytic bacteria and urinary excretion of purine derivatives in growing beef steers. Anim Feed Sci. Tech. 2016, 221, 185–194. [Google Scholar] [CrossRef]
- Fondevila, M.; Dehority, B.A. Interactions between Fibrobacter succinogenes, Prevotella ruminicola, and Ruminococcus flavefaciens in the digestion of cellulose from forages. J. Anim Sci. 1996, 74, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, T.H.; Hungate, R.E. Succinic acid turnover and propionate production in the bovine rumen. Appl. Environ. Microbiol. 1963, 11, 132–135. [Google Scholar] [CrossRef]
- Cieslak, A.; Zmora, P.; Perskamczyc, E.; Szumacherstrabel, M. Effects of tannins source (Vaccinium vitis idaea L.) on rumen microbial fermentation in vivo. Anim. Feed Sci. Tech. 2012, 176, 102–106. [Google Scholar] [CrossRef]
- Hino, T.; Kuroda, S. Presence of lactate dehydrogenase and lactate racemase in megasphaera elsdenii grown on glucose or lactate. Appl. Environ. Microbiol. 1993, 59, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Soest, P.J.V. Nutritional Ecology of the Ruminant; Cornell university press: New York, NY, USA, 2018; Volume 44, pp. 2552–2561. [Google Scholar]
- Kristensen, N.B. Rumen microbial sequestration of [2-(13)C] acetate in cattle. J. Anim Sci. 2001, 79, 2491–2498. [Google Scholar] [CrossRef]
- Hackmann, T.J.; Firkins, J.L. Maximizing efficiency of rumen microbial protein production. Front. Microbiol. 2015, 6, 465–471. [Google Scholar] [CrossRef]
- Diezgonzalez, F.; Bond, D.R.; Jennings, E.; Russell, J.B. Alternative schemes of butyrate production in Butyrivibrio fibrisolvens and their relationship to acetate utilization, lactate production, and phylogeny. Arch. Microbiol. 1999, 171, 324–330. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | HF | HC | SEM 5 | p-Value 6 | ||||
|---|---|---|---|---|---|---|---|---|
| BF0h 3 | AF4h 4 | BF0 h | AF4 h | Feed | Time | Feed × Time | ||
| pH | 6.90 | 6.06 | 6.84 | 5.76 | 0.01 | NS | ** | * |
| Acetate, mM | 79.84 | 111.69 | 69.24 | 92.94 | 24.70 | ** | ** | NS |
| Propionate,mM | 19.32 | 23.87 | 27.12 | 49.20 | 5.76 | ** | ** | ** |
| Butyrate, mM | 9.73 | 13.56 | 10.51 | 18.76 | 3.16 | ** | ** | * |
| TVFA 1, mM | 108.89 | 149.12 | 106.87 | 160.90 | 52.97 | ** | ** | NS |
| A/P 2 | 4.21 | 4.67 | 2.56 | 1.91 | 0.04 | ** | NS | ** |
| Lactate (mmol/L) | 7.27 | 5.40 | 8.48 | 6.61 | 0.86 | NS | ** | NS |
| Pyruvate (μg/L) | 263.46 | 290.68 | 313.60 | 282.49 | 131.15 | * | NS | * |
| Enzyme | HF | HC | SEM 3 | p-Value 4 | ||||
|---|---|---|---|---|---|---|---|---|
| BF0h 1 | AF4h 2 | BF0h | AF4h | Feed | Time | Feed × Time | ||
| EC:6.4.1.1 | 3.1801 | 3.6213 | 2.7825 | 2.6925 | 0.0456 | ** | NS | NS |
| EC:1.1.1.37 | 2.6938 | 3.3037 | 2.8694 | 3.3698 | 0.0712 | NS | * | NS |
| EC:4.2.1.2 | 3.5578 | 4.1535 | 3.6002 | 3.7724 | 0.1473 | NS | NS | NS |
| EC: 1.3.5.1 | 4.8624 | 5.7332 | 4.7385 | 4.8726 | 0.1725 | NS | NS | NS |
| EC:6.2.1.5 | 0.1001 | 0.1617 | 1.0779 | 1.5528 | 0.0212 | ** | * | NS |
| EC:5.4.99.2 | 4.8603 | 5.5625 | 4.8684 | 5.1543 | 0.0604 | NS | * | NS |
| EC:6.4.1.3 | 3.4469 | 3.9384 | 3.4114 | 3.3103 | 0.0303 | NS | NS | NS |
| EC:2.8.3.1 | 0.0161 | 0.0313 | 0.0022 | 0.0664 | 0.0002 | * | ** | ** |
| Enzyme | HF | HC | SEM 3 | p-Value 4 | ||||
|---|---|---|---|---|---|---|---|---|
| BF0h 1 | AF4h 2 | BF0h | AF4h | Feed | Time | Feed × Time | ||
| EC: 1.2.7.1 | 0.0197 | 0.0144 | 0.5551 | 1.0831 | 0.0201 | ** | * | * |
| EC:2.3.1.9 | 0.2156 | 0.4360 | 0.1077 | 0.4766 | 0.0047 | NS | * | NS |
| EC:1.1.1.157 | 0.4243 | 0.4258 | 0.3754 | 0.3273 | 0.0054 | NS | NS | NS |
| EC:4.2.1.17 | 0.2757 | 0.2754 | 0.3051 | 0.2771 | 0.0033 | NS | NS | NS |
| EC:1.3.8.1 | 0.8092 | 0.7743 | 0.2756 | 0.2281 | 0.0075 | ** | NS | NS |
| EC:2.8.3.8 | 0.4289 | 0.4697 | 0.3150 | 0.3494 | 0.0038 | * | NS | NS |
| EC:4.2.1.55 | 0.0601 | 0.1067 | 0.0795 | 0.0992 | 0.0000 | NS | ** | * |
| EC:2.3.1.19 | 2.0545 | 2.2858 | 1.8426 | 2.0529 | 0.0127 | * | * | NS |
| EC:2.7.2.7 | 1.6690 | 2.0296 | 2.1763 | 2.4183 | 0.0148 | ** | * | NS |
| Bacteria | HF | HC | SEM 3 | p-Value 4 | ||||
|---|---|---|---|---|---|---|---|---|
| BF0h 1 | AF4h 2 | BF0h | AF4h | Feed | Time | Feed × Time | ||
| Ruminococcus flavefaciens | 1.9712 | 2.6007 | 2.3312 | 2.6435 | 0.0421 | NS | * | NS |
| Ruminococcus albus | 0.1562 | 0.3060 | 0.3612 | 0.2482 | 0.0021 | NS | NS | ** |
| Fibrobacter succinogenes | 0.3152 | 0.5240 | 0.1860 | 0.3638 | 0.0017 | ** | ** | NS |
| Butyrivibrio fibrisolvens | 0.0388 | 0.0180 | 0.0557 | 0.0672 | 0.000 | ** | NS | ** |
| Prevotella ruminicola | 3.8373 | 2.2827 | 2.9234 | 5.0617 | 0.0937 | ** | NS | ** |
| Selenomonas ruminantium | 0.8129 | 1.2319 | 0.6473 | 5.5426 | 0.0380 | ** | ** | ** |
| Megasphaera elsdenii | 0.0099 | 0.0148 | 0.0022 | 0.0033 | 3.1E-6 | ** | NS | NS |
| Veillonella alkalescens | 0.0140 | 0.0885 | 0.2692 | 0.1966 | 0.0010 | ** | NS | * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhang, G.; Li, Y.; Zhang, Y. Effects of High Forage/Concentrate Diet on Volatile Fatty Acid Production and the Microorganisms Involved in VFA Production in Cow Rumen. Animals 2020, 10, 223. https://doi.org/10.3390/ani10020223
Wang L, Zhang G, Li Y, Zhang Y. Effects of High Forage/Concentrate Diet on Volatile Fatty Acid Production and the Microorganisms Involved in VFA Production in Cow Rumen. Animals. 2020; 10(2):223. https://doi.org/10.3390/ani10020223
Chicago/Turabian StyleWang, Lijun, Guangning Zhang, Yang Li, and Yonggen Zhang. 2020. "Effects of High Forage/Concentrate Diet on Volatile Fatty Acid Production and the Microorganisms Involved in VFA Production in Cow Rumen" Animals 10, no. 2: 223. https://doi.org/10.3390/ani10020223
APA StyleWang, L., Zhang, G., Li, Y., & Zhang, Y. (2020). Effects of High Forage/Concentrate Diet on Volatile Fatty Acid Production and the Microorganisms Involved in VFA Production in Cow Rumen. Animals, 10(2), 223. https://doi.org/10.3390/ani10020223