Insights into Genetic Diversity, Runs of Homozygosity and Heterozygosity-Rich Regions in Maremmana Semi-Feral Cattle Using Pedigree and Genomic Data

 ,
,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples, Genotyping and Data Editing
2.2. Genetic Diversity Coefficients
2.3. Runs of Homozygosity Detection and Distribution
2.4. Sensitivity Analysis
2.5. Detection of Heterozygosity-Rich Regions
2.6. Genealogical Analysis
2.7. SNP-Based and ROH-Based Genomic Relationships
2.8. Gene Annotation
3. Results
3.1. Genetic Diversity
3.2. Pedigree and Genomic Relationships and Inbreeding Coefficients
3.3. Runs of Homozygosity
3.4. Heterozygosity-Rich Regions
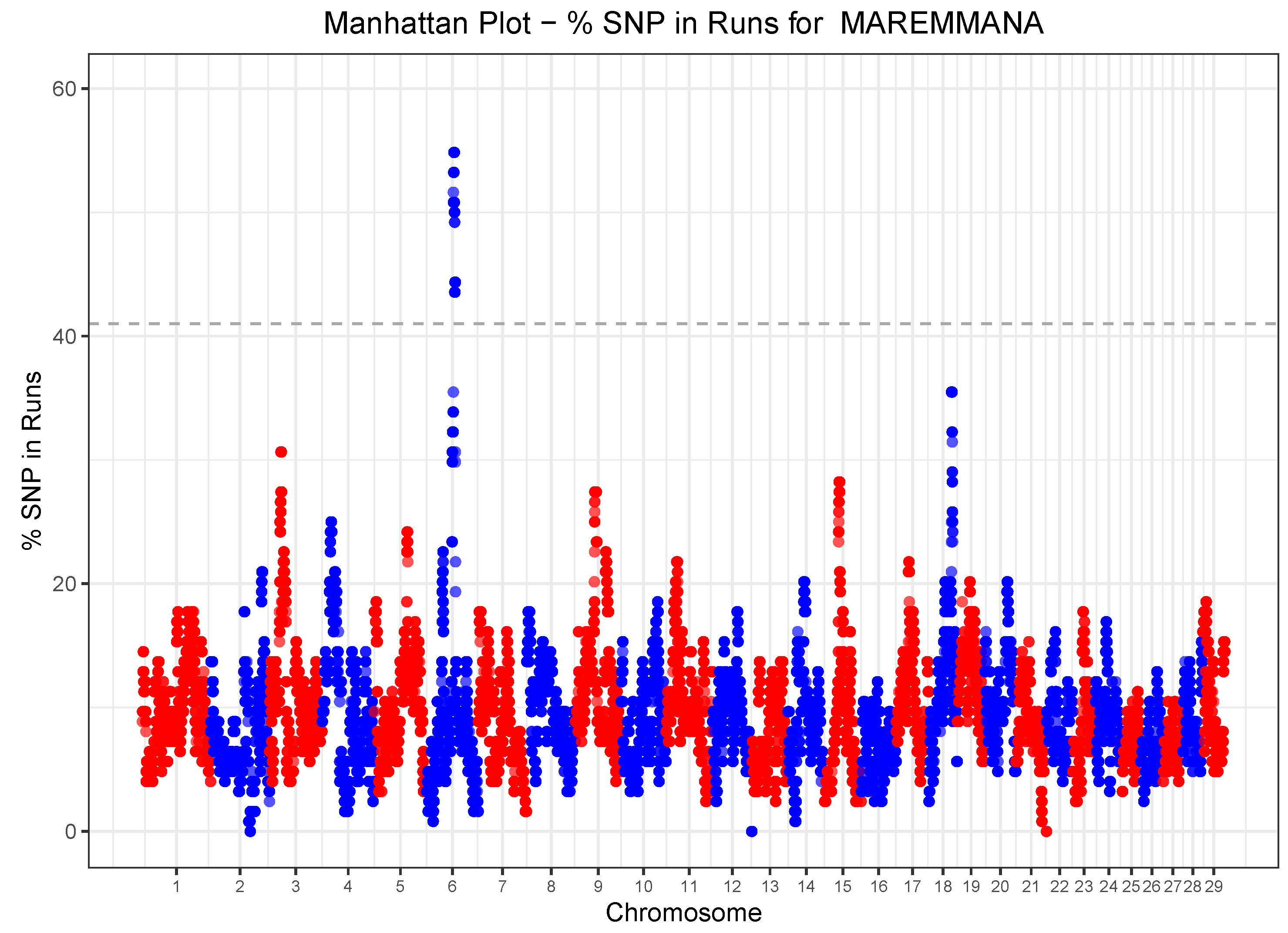
3.5. Runs of Homozygosity (ROH) and Heterozygosity (ROHet) Islands
3.6. ROH Sensitivity Analysis
4. Discussion
4.1. Genetic Diversity of Maremmana Cattle
4.2. Runs of Homozygosity and Heterozygosity-Rich Regions
4.3. Biological Meaning of ROH-Islands and Islands of Heterozygosity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kugler, W.; Broxham, E. The ecological value of feral livestock populations in Europe. In Overview, Situation and Development of a Network for Management of Wild Livestock Populations; Final Report, SAVE-Project; SAVE Foundation: St. Gallen, Switzerland, 2014. [Google Scholar]
- Bruford, M.W.; Ginja, C.; Hoffmann, I.; Joost, S.; Orozco-terWengel, P.; Alberto, F.J.; Amaral, A.J.; Barbato, M.; Biscarini, F.; Colli, L.; et al. Prospects and challenges for the conservation of farm animal genomic resources, 2015–2025. Front. Genet. 2015, 6, 314. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Mészáros, G.; Boison, S.A.; Pérez O’Brien, A.M.; Ferenčaković, M.; Curik, I.; Da Silva, M.V.B.; Utsunomiya, Y.T.; Garcia, J.F.; Sölkner, J. Genomic analysis for managing small and endangered populations: A case study in Tyrol Grey cattle. Front. Genet. 2015, 6, 173. [Google Scholar] [PubMed]
- Gurgul, A.; Szmatoła, T.; Topolski, P.; Jasielczuk, I.; Żukowski, K.; Bugno-Poniewierska, M. The use of runs of homozygosity for estimation of recent inbreeding in Holstein cattle. J. Appl. Genet. 2016, 57, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Tolone, M.; Di Gerlando, R.; Fontanesi, L.; Sardina, M.; Portolano, B. Genomic inbreeding estimation in small populations: Evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 2016, 10, 746–754. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Ferenčaković, M.; Hamzić, E.; Gredler, B.; Solberg, T.; Klemetsdal, G.; Curik, I.; Sölkner, J. Estimates of autozygosity derived from runs of homozygosity: Empirical evidence from selected cattle populations. J. Anim. Breed. Genet. 2013, 130, 286–293. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Metzger, J.; Karwath, M.; Tonda, R.; Beltran, S.; Águeda, L.; Gut, M.; Gut, I.G.; Distl, O. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genom. 2015, 16, 764. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Bosse, M.; Lund, M.S.; Sahana, G. Runs of homozygosity and distribution of functional variants in the cattle genome. BMC Genom. 2015, 16, 542. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; McParland, S.; Wall, E.; Berry, D.P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE 2017, 12, e0176780. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Sardina, M.; Tolone, M.; Di Gerlando, R.; Sutera, A.; Fontanesi, L.; Portolano, B. Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds. Anim. Int. J. Anim. Biosci. 2018, 12, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Bovo, S.; Bertolini, F.; Dall’Olio, S.; Costa, L.N.; Tinarelli, S.; Gallo, M.; Fontanesi, L. Runs of homozygosity islands in Italian cosmopolitan and autochthonous pig breeds identify selection signatures in the porcine genome. Livest. Sci. 2020, 240, 104219. [Google Scholar] [CrossRef]
- Williams, J.L.; Hall, S.J.; Del Corvo, M.; Ballingall, K.; Colli, L.; Ajmone Marsan, P.; Biscarini, F. Inbreeding and purging at the genomic Level: The Chillingham cattle reveal extensive, non-random SNP heterozygosity. Anim. Genet. 2016, 47, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Bizarria dos Santos, W.; Pimenta Schettini, G.; Fonseca, M.G.; Pereira, G.L.; Loyola Chardulo, L.A.; Rodrigues Machado Neto, O.; Baldassini, W.A.; Nunes de Oliveira, H.; Abdallah Curi, R. Fine-scale estimation of inbreeding rates, runs of homozygosity and genome-wide heterozygosity levels in the Mangalarga Marchador horse breed. J. Anim. Breed. Genet. 2020. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Catillo, G.; Moioli, B.; Napolitano, F.; Steri, R. Identification of genomic regions harboring diversity between Holstein and two local endangered breeds, Modenese and Maremmana. Livest. Sci. 2018, 216, 75–83. [Google Scholar] [CrossRef]
- Flori, L.; Moazami-Goudarzi, K.; Alary, V.; Araba, A.; Boujenane, I.; Boushaba, N.; Casabianca, F.; Casu, S.; Ciampolini, R.; Coeur D’Acier, A.; et al. A genomic map of climate adaptation in Mediterranean cattle breeds. Mol. Ecol. 2019, 28, 1009–1029. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Do, C. LDNE: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 2008, 8, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Doekes, H.P.; Veerkamp, R.F.; Bijma, P.; de Jong, G.; Hiemstra, S.J.; Windig, J.J. Inbreeding depression due to recent and ancient inbreeding in Dutch Holstein–Friesian dairy cattle. Genet. Sel. Evol. 2019, 51, 54. [Google Scholar] [CrossRef] [PubMed]
- Makanjuola, B.O.; Maltecca, C.; Miglior, F.; Schenkel, F.S.; Baes, C.F. Effect of recent and ancient inbreeding on production and fertility traits in Canadian Holsteins. BMC Genom. 2020, 21, 1–15. [Google Scholar] [CrossRef]
- Peripolli, E.; Munari, D.; Silva, M.; Lima, A.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. R package version 0.9. 5. 2018. Available online: https://cran.r-project.org/web/packages/detectRUNS/index.html (accessed on 2 December 2020).
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsan, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2015, 46, 110–121. [Google Scholar] [CrossRef]
- Mrode, R.A. Linear Models for the Prediction of Animal Breeding Values; Cabi: Wallingford, CO, USA, 2014. [Google Scholar]
- Wright, S. Coefficients of inbreeding and relationship. Am. Nat. 1922, 56, 330–338. [Google Scholar] [CrossRef]
- Coster, A.; Coster, M.A. Package ‘pedigree’. R Package Version 1.4. 2010. Available online: https://cran.r-project.org/web/packages/pedigree/index.html (accessed on 2 December 2020).
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef]
- Perez Torrecillas, C.; Bozzi, R.; Negrini, R.; Filippini, F.; Giorgetti, A. Genetic variability of three Italian cattle breeds determined by parameters based on probabilities of gene origin. J. Anim. Breed. Genet. 2002, 119, 274–279. [Google Scholar] [CrossRef]
- Fioretti, M.; Negrini, R.; Biffani, S.; Quaglia, A.; Valentini, A.; Nardone, A. Demographic structure and population dynamics of Maremmana cattle local breed after 35 years of traditional selection. Livest. Sci. 2020, 232, 103903. [Google Scholar] [CrossRef]
- Sved, J. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Orozco-terWengel, P.; Barbato, M.; Nicolazzi, E.; Biscarini, F.; Milanesi, M.; Davies, W.; Williams, D.; Stella, A.; Ajmone-Marsan, P.; Bruford, M.W. Revisiting demographic processes in cattle with genome-wide population genetic analysis. Front. Genet. 2015, 6, 191. [Google Scholar] [CrossRef] [PubMed]
- Zsolnai, A.; Maróti-Agóts, Á.; Kovács, A.; Bâlteanu, A.; Kaltenecker, E.; Anton, I. Genetic position of Hungarian Grey among European cattle and identification of breed-specific markers. Animal 2020, 14, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Senczuk, G.; Mastrangelo, S.; Marsan, P.A.; Becskei, Z.; Colangelo, P.; Colli, L.; Ferretti, L.; Karsli, T.; Lancioni, H.; Lasagna, E.; et al. Into the origin and diversification of Podolian cattle breeds by testing Neolithic scenarios of European colonization using genome-wide SNP data. Authorea Prepr. 2020. [Google Scholar] [CrossRef]
- Moioli, B.; Napolitano, F.; Catillo, G. Genetic diversity between Piedmontese, Maremmana, and Podolica cattle breeds. J. Hered. 2004, 95, 250–256. [Google Scholar] [CrossRef]
- Negrini, R.; Milanesi, E.; Bozzi, R.; Pellecchia, M.; Ajmone-Marsan, P. Tuscany autochthonous cattle breeds: An original genetic resource investigated by AFLP markers. J. Anim. Breed. Genet. 2006, 123, 10–16. [Google Scholar] [CrossRef]
- Maretto, F.; Ramljak, J.; Sbarra, F.; Penasa, M.; Mantovani, R.; Ivanković, A.; Bittante, G. Genetic relationships among Italian and Croatian Podolian cattle breeds assessed by microsatellite markers. Livest. Sci. 2012, 150, 256–264. [Google Scholar] [CrossRef]
- Bozzi, R.; Sirtori, F.; Forabosco, F.; Franci, O. Pedigree analysis of Italian beef cattle breeds. Ital. J. Anim. Sci. 2003, 2, 121–123. [Google Scholar]
- Zhang, Y.; Young, J.; Wang, C.; Sun, X.; Wolc, A.; Dekkers, J. Inbreeding by pedigree and genomic markers in selection lines of pigs. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar]
- Szmatoła, T.; Gurgul, A.; Jasielczuk, I.; Ząbek, T.; Ropka-Molik, K.; Litwińczuk, Z.; Bugno-Poniewierska, M. A comprehensive analysis of runs of homozygosity of eleven cattle breeds representing different production types. Animals 2019, 9, 1024. [Google Scholar] [CrossRef] [PubMed]
- Szmatoła, T.; Gurgul, A.; Ropka-Molik, K.; Jasielczuk, I.; Ząbek, T.; Bugno-Poniewierska, M. Characteristics of runs of homozygosity in selected cattle breeds maintained in Poland. Livest. Sci. 2016, 188, 72–80. [Google Scholar] [CrossRef]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genom. 2011, 12, 460. [Google Scholar] [CrossRef] [PubMed]
- Ferenčaković, M.; Banadinović, M.; Mercvajler, M.; Khayatzadeh, N.; Mészáros, G.; Cubric-Curik, V.; Curik, I.; Söòkner, J. Mapping of heterozygosity rich regions in Austrian pinzgauer cattle. Acta Agric. Slov. 2016, 5, 42. [Google Scholar]
- Marras, G.; Wood, B.; Makanjuola, B.; Malchiodi, F.; Peeters, K.; van As, P.; Baes, C.; Biscarini, F. Characterization of runs of homozygosity and heterozygosity-rich regions in a commercial turkey (Meleagris gallopavo) population. In Proceedings of the 11th World Congress of Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018. [Google Scholar]
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef] [PubMed]
- Forutan, M.; Mahyari, S.A.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, G.; Yang, L.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; Li, J. Genomic patterns of homozygosity in Chinese local cattle. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Szmatoła, T.; Jasielczuk, I.; Semik-Gurgul, E.; Szyndler-Nędza, M.; Blicharski, T.; Szulc, K.; Skrzypczak, E.; Gurgul, A. Detection of runs of homozygosity in conserved and commercial pig breeds in Poland. J. Anim. Breed. Genet. 2020. [Google Scholar] [CrossRef]
- Hayes, B.J.; Lien, S.; Nilsen, H.; Olsen, H.G.; Berg, P.; MacEachern, S.; Potter, S.; Meuwissen, T. The origin of selection signatures on bovine chromosome 6. Anim. Genet. 2008, 39, 105–111. [Google Scholar] [CrossRef]
- Hay, E.H.; Roberts, A. Genotype× prenatal and post-weaning nutritional environment interaction in a composite beef cattle breed using reaction norms and a multi-trait model. J. Anim. Sci. 2018, 96, 444–453. [Google Scholar] [CrossRef]
- Ouyang, H.; Zhang, H.; Li, W.; Liang, S.; Jebessa, E.; Abdalla, B.A.; Nie, Q. Identification, expression and variation of the GNPDA2 gene, and its association with body weight and fatness traits in chicken. PeerJ 2016, 4, e2129. [Google Scholar] [CrossRef] [PubMed]
- Mc Parland, S.; Kearney, F.; Berry, D.P. Purging of inbreeding depression within the Irish Holstein-Friesian population. Genet. Sel. Evol. 2009, 41, 16. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.; Olson, K.; Wiggans, G.; Cole, J.; Tooker, M. Genomic inbreeding and relationships among Holsteins, Jerseys, and Brown Swiss. J. Dairy Sci. 2011, 94, 5673–5682. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Matukumalli, L.K.; Li, C.; Song, J.; Gasbarre, L.C.; Van Tassell, C.P.; Sonstegard, T.S. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct. Integr. Genom. 2012, 12, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A.; Jiang, Y.; et al. Single step genome-wide association studies based on genotyping by sequence data reveals novel loci for the litter traits of domestic pigs. Genomics 2018, 110, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.O.; Mekonnen, Y.A.; Gültas, M.; Effa, K.; Hanotte, O. Identification of candidate signature genes and key regulators associated with Trypanotolerance in the Sheko Breed. Front. Genet. 2019, 10, 1095. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class (MB) | N. | Avg. Size (kB) | Freq. |
|---|---|---|---|
| 2–4 | 1206 | 2998 | 0.36 |
| 4–8 | 1053 | 5607 | 0.32 |
| 8–12 | 416 | 9737 | 0.12 |
| 12–16 | 211 | 13,852 | 0.06 |
| 16–20 | 153 | 17,786 | 0.05 |
| 20–30 | 162 | 24,085 | 0.05 |
| >30 | 131 | 40,972 | 0.04 |
| Class (kB) | N. | Avg. Size (kB) | Freq. |
|---|---|---|---|
| 250–500 | 165 | 451 | 0.11 |
| 500–750 | 821 | 617 | 0.56 |
| 750–1000 | 372 | 850 | 0.25 |
| 1000–1500 | 106 | 1141 | 0.07 |
| 1500–2000 | 5 | 1744 | 0.00 |
| >2000 | 2 | 2071 | 0.00 |
| BTA | Start (bp) | End (bp) | N SNPs | Genes | |
|---|---|---|---|---|---|
| ROH islands | 6 | 61,877,121 | 64,865,529 | 58 | LOC112447156, LOC112447157, LOC101906152, TRNAC-ACA, KCTD8, YIPF7, GUF1, GNPDA2, TRNAC-ACA, LOC112447181, LOC112447015, GABRG1, GABRA2 |
| 6 | 65,343,631 | 65,774,333 | 15 | COX7B2, GABRA4, GABRB1 | |
| ROHet islands | 14 | 51,026,484 | 51,596,222 | 16 | LOC112449555, LOC112449527 |
| 21 | 28,714,736 | 29,509,054 | 18 | LOC104975368, TARSL2, TM2D3, PCSK6, LOC101906554, TRNAC-GCA, LOC100300380, SNRPA1, LOC100848886, LOC789895, LOC112443140, LOC112443139 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biscarini, F.; Mastrangelo, S.; Catillo, G.; Senczuk, G.; Ciampolini, R. Insights into Genetic Diversity, Runs of Homozygosity and Heterozygosity-Rich Regions in Maremmana Semi-Feral Cattle Using Pedigree and Genomic Data. Animals 2020, 10, 2285. https://doi.org/10.3390/ani10122285
Biscarini F, Mastrangelo S, Catillo G, Senczuk G, Ciampolini R. Insights into Genetic Diversity, Runs of Homozygosity and Heterozygosity-Rich Regions in Maremmana Semi-Feral Cattle Using Pedigree and Genomic Data. Animals. 2020; 10(12):2285. https://doi.org/10.3390/ani10122285
Chicago/Turabian StyleBiscarini, Filippo, Salvatore Mastrangelo, Gennaro Catillo, Gabriele Senczuk, and Roberta Ciampolini. 2020. "Insights into Genetic Diversity, Runs of Homozygosity and Heterozygosity-Rich Regions in Maremmana Semi-Feral Cattle Using Pedigree and Genomic Data" Animals 10, no. 12: 2285. https://doi.org/10.3390/ani10122285
APA StyleBiscarini, F., Mastrangelo, S., Catillo, G., Senczuk, G., & Ciampolini, R. (2020). Insights into Genetic Diversity, Runs of Homozygosity and Heterozygosity-Rich Regions in Maremmana Semi-Feral Cattle Using Pedigree and Genomic Data. Animals, 10(12), 2285. https://doi.org/10.3390/ani10122285