Microbiome Shift, Diversity, and Overabundance of Opportunistic Pathogens in Bovine Digital Dermatitis Revealed by 16S rRNA Amplicon Sequencing

 ,
, {kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Metagenomics Sequencing and Diversity Analysis
2.3. Divergence and Phylogenetic Analysis
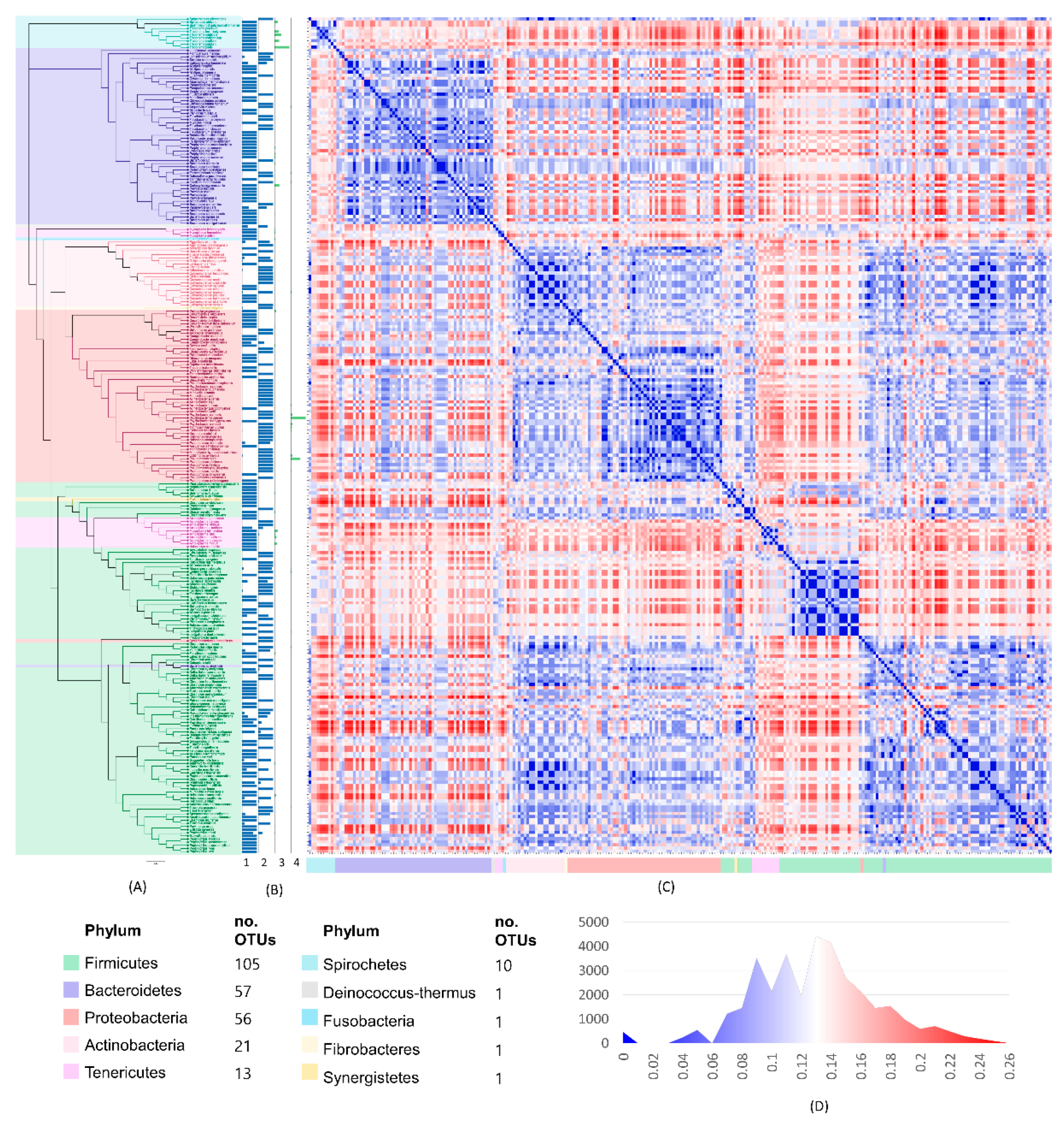
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Evans, N.J.; Murray, R.D.; Carter, S.D. Bovine digital dermatitis: Current concepts from laboratory to farm. Vet. J. 2016, 211, 3–13. [Google Scholar] [CrossRef]
- Demirkan, I.; Evans, N.J.; Singh, P.; Brown, J.M.; Getty, B.; Carter, S.D.; Timofte, D.; Hart, C.A.; Vink, W.D.; Birtles, R.J.; et al. Association of unique, isolated treponemes with bovine digital dermatitis lesions. J. Clin. Microbiol. 2009, 47, 689–696. [Google Scholar] [CrossRef]
- Klitgaard, K.; Nielsen, M.W.; Ingerslev, H.C.; Boye, M.; Jensen, T.K. Discovery of bovine digital dermatitis-associated Treponema spp. in the dairy herd environment by a targeted deep-sequencing approach. Appl. Environ. Microbiol. 2014, 80, 4427–4432. [Google Scholar] [CrossRef] [PubMed]
- De Jesús Argáez-Rodríguez, F.; Hird, D.W.; Hernández De Anda, J.; Read, D.H.; Rodríguez-Lainz, A. Papillomatous digital dermatitis on a commercial dairy farm in Mexicali, Mexico: Incidence and effect on reproduction and milk production. Prev. Vet. Med. 1997, 32, 275–286. [Google Scholar] [CrossRef]
- Read, D.H.; Walker, R.L. Papillomatous digital dermatitis (footwarts) in California dairy cattle: Clinical and gross pathologic findings. J. Vet. Diagn. Investig. 1998, 10, 67–76. [Google Scholar] [CrossRef]
- Elliott, M.K.; Alt, D.P.; Zuerner, R.L. Lesion formation and antibody response induced by papillomatous digital dermatitis-associated spirochetes in a murine abscess model. Infect. Immun. 2007, 75, 4400–4408. [Google Scholar] [CrossRef] [PubMed]
- Cheli, R.; Mortellaro, C. Digital dermatitis in cattle. In Proceedings of the 8th International Conference Diseases of Cattle, Milan, Italy, 9–13 September 1974; pp. 208–213. [Google Scholar]
- Palmer, M.A.; O’Connell, N.E. Digital dermatitis in dairy cows: A review of risk factors and potential sources of between-animal variation in susceptibility. Animals 2015, 5, 512–535. [Google Scholar] [CrossRef]
- Nally, J.E.; Hornsby, R.L.; Alt, D.P.; Whitelegge, J.P. Phenotypic and proteomic characterization of treponemes associated with bovine digital dermatitis. Vet. Microbiol. 2019, 235, 35–42. [Google Scholar] [CrossRef]
- Wilson-Welder, J.H.; Alt, D.P.; Nally, J.E. Digital dermatitis in cattle: Current bacterial and immunological findings. Animals 2015, 5, 1114–1135. [Google Scholar] [CrossRef]
- Zinicola, M.; Lima, F.; Lima, S.; Machado, V.; Gomez, M. Altered microbiomes in bovine digital dermatitis lesions, and the gut as a pathogen reservoir. PLoS ONE 2015, 1–23. [Google Scholar] [CrossRef]
- Nielsen, M.W.; Strube, M.L.; Isbrand, A.; Al-Medrasi, W.D.H.M.; Boye, M.; Jensen, T.K.; Klitgaard, K. Potential bacterial core species associated with digital dermatitis in cattle herds identified by molecular profiling of interdigital skin samples. Vet. Microbiol. 2016, 186, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Krull, A.C.; Shearer, J.K.; Gorden, P.J.; Cooper, V.L.; Phillips, G.J.; Plummera, P.J. Deep sequencing analysis reveals temporal microbiota changes associated with development of bovine digital dermatitis. Infect. Immun. 2014, 82, 3359–3373. [Google Scholar] [CrossRef] [PubMed]
- Moreira, T.F.; Facury Filho, E.J.; Carvalho, A.U.; Strube, M.L.; Nielsen, M.W.; Klitgaard, K.; Jensen, T.K. Pathology and bacteria related to digital dermatitis in dairy cattle in all year round grazing system in Brazil. PLoS ONE 2018, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hesseling, J.; Legione, A.R.; Stevenson, M.A.; McCowan, C.I.; Pyman, M.F.; Finochio, C.; Nguyen, D.; Roic, C.L.; Thiris, O.L.; Zhang, A.J.; et al. Bovine digital dermatitis in Victoria, Australia. Aust. Vet. J. 2019, 97, 404–413. [Google Scholar] [CrossRef]
- Mamuad, L.L.; Joo, B.; Al, S.; Espiritu, H.M.; Jeong, S.; Kim, W.; Lee, S.; Cho, Y. Treponema spp., the dominant pathogen in the lesion of bovine digital dermatitis and its characterization in dairy cattle. Vet. Microbiol. 2020, 245, 108696. [Google Scholar] [CrossRef]
- Zinicola, M.; Higgins, H.; Lima, S.; Machado, V.; Guard, C.; Bicalho, R. Shotgun metagenomic sequencing reveals functional genes and microbiome associated with bovine digital dermatitis. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high- throughput community sequencing data intensity normalization improves color calling in SOLiD sequencing. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, L.; Gu, Y.Q.; Coleman-Derr, D. MetaCoMET: A web platform for discovery and visualization of the core microbiome. Bioinformatics 2016, 32, 3469–3470. [Google Scholar] [CrossRef]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. Gigascience 2012, 464, 1–6. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl. Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.M.; Harrison, A.O.; McAllister, S.M.; Polson, S.W.; Wommack, K.E. Iroki: Automatic customization and visualization of phylogenetic trees. PeerJ 2020, 8, e8584. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucl. Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Beninger, C.; Naqvi, S.A.; Naushad, S.; Orsel, K.; Luby, C.; Derakhshani, H.; Khafipour, E.; De Buck, J. Associations between digital dermatitis lesion grades in dairy cattle and the quantities of four Treponema species. Vet. Res. 2018, 49, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Bay, V.; Griffiths, B.; Carter, S.; Evans, N.J.; Lenzi, L.; Bicalho, R.C.; Oikonomou, G. 16S rRNA amplicon sequencing reveals a polymicrobial nature of complicated claw horn disruption lesions and interdigital phlegmon in dairy cattle. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Yang, S.H.; Seo, H.S.; Woo, J.H.; Oh, H.M.; Jang, H.; Lee, J.H.; Kim, S.J.; Kwon, K.K. Carboxylicivirga gen. nov. in the family Marinilabiliaceae with two novel species, Carboxylicivirga mesophila sp. nov. and Carboxylicivirga taeanensis sp. nov., and reclassification of Cytophaga fermentans as Saccharicrinis fermentans gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 1351–1358. [Google Scholar] [CrossRef]
- Belkacemi, S.; Khalil, J.B.; Ominami, Y.; Hisada, A.; Fontanini, A.; Caputo, A.; Levasseur, A.; Lagier, J.C.; Khelaifia, S.; Raoult, D. Passive filtration, rapid scanning electron microscopy, and matrix-assisted laser desorption ionization–time of flight mass spectrometry for Treponema culture and identification from the oral cavity. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef]
- Yano, T.; Moe, K.K.; Yamazaki, K.; Ooka, T.; Hayashi, T.; Misawa, N. Identification of candidate pathogens of papillomatous digital dermatitis in dairy cattle from quantitative 16S rRNA clonal analysis. Vet. Microbiol. 2010, 143, 352–362. [Google Scholar] [CrossRef]
- Santos, T.M.A.; Pereira, R.V.; Caixeta, L.S.; Guard, C.L.; Bicalho, R.C. Microbial diversity in bovine Papillomatous digital dermatitis in Holstein dairy cows from upstate New York. FEMS Microbiol. Ecol. 2012, 79, 518–529. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espiritu, H.M.; Mamuad, L.L.; Kim, S.-h.; Jin, S.-j.; Lee, S.-s.; Kwon, S.-w.; Cho, Y.-i. Microbiome Shift, Diversity, and Overabundance of Opportunistic Pathogens in Bovine Digital Dermatitis Revealed by 16S rRNA Amplicon Sequencing. Animals 2020, 10, 1798. https://doi.org/10.3390/ani10101798
Espiritu HM, Mamuad LL, Kim S-h, Jin S-j, Lee S-s, Kwon S-w, Cho Y-i. Microbiome Shift, Diversity, and Overabundance of Opportunistic Pathogens in Bovine Digital Dermatitis Revealed by 16S rRNA Amplicon Sequencing. Animals. 2020; 10(10):1798. https://doi.org/10.3390/ani10101798
Chicago/Turabian StyleEspiritu, Hector M., Lovelia L. Mamuad, Seon-ho Kim, Su-jeong Jin, Sang-suk Lee, Seok-won Kwon, and Yong-il Cho. 2020. "Microbiome Shift, Diversity, and Overabundance of Opportunistic Pathogens in Bovine Digital Dermatitis Revealed by 16S rRNA Amplicon Sequencing" Animals 10, no. 10: 1798. https://doi.org/10.3390/ani10101798
APA StyleEspiritu, H. M., Mamuad, L. L., Kim, S.-h., Jin, S.-j., Lee, S.-s., Kwon, S.-w., & Cho, Y.-i. (2020). Microbiome Shift, Diversity, and Overabundance of Opportunistic Pathogens in Bovine Digital Dermatitis Revealed by 16S rRNA Amplicon Sequencing. Animals, 10(10), 1798. https://doi.org/10.3390/ani10101798