Genetics of Arthrogryposis and Macroglossia in Piemontese Cattle Breed

 , , ,
, , ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals, Genotyping, and Data Editing
2.3. Statistical Analysis
2.4. Candidate Gene Detection
3. Results
3.1. Genome-wide Association Study and FST
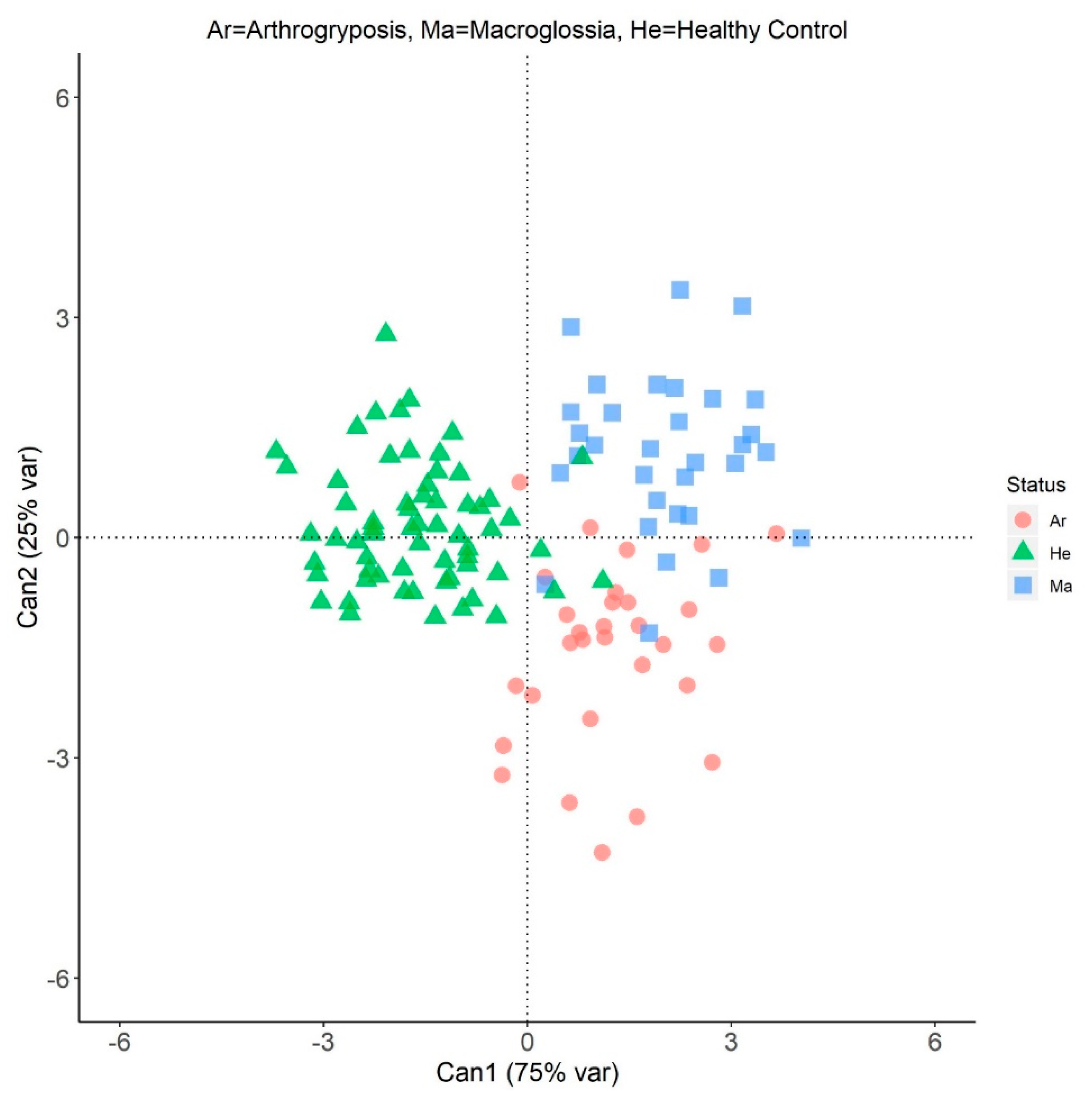
3.2. Canonical Discriminant Analysis
3.3. Candidate Gene Detection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hutt, F.B. A hereditary lethal muscle contracture in cattle. J. Hered. 1934, 25, 41–46. [Google Scholar] [CrossRef]
- Leipold, H.W.; Cates, W.F.; Radostis, O.M.; Howell, W.E. Arthrogryposis and associated defects in newborn calves. Am. J. Vet. Res. 1970, 31, 1367–1374. [Google Scholar] [PubMed]
- Huston, K.; Saperstein, G.; Steffen, D.; Millar, P.; Lauvergne, J.J. Clinical, pathological and other visible traits loci except coat colour (category 2). In Mendelian Inheritance in Cattle 2000; Millar, P., Lauvergne, J.J., Dolling, C., Eds.; EAAP Publication: Wageningen, The Netherlands, 2000; Volume 101, pp. 164–175. [Google Scholar]
- Shupe, J.L.; Binns, W.; James, L.F.; Keeler, R.F. Lupine, a cause of crooked calf disease. J. Am. Vet. Med. Assoc. 1967, 151, 198–203. [Google Scholar]
- Anderson, D.E.; Desrochers, A.; St. Jean, G. Management of Tendon Disorders in Cattle. Vet. Clin. N. Am. Food Anim. Pract. 2008, 24, 551–566. [Google Scholar] [CrossRef]
- Lauvergne, J.J.; Vissac, B.; Perramon, A. Étude du caractère culard. I. Mise au point bibliographique. Annales de zootechnie, INRA/EDP. Sciences 1963, 12, 133–156. [Google Scholar]
- Webb, E.C.; Casey, N.H. Physiological limits to growth and the related effects on meat quality. Livest. Sci. 2010, 130, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Best, L.G.; Gilbert-Barness, E.; Gerrard, D.E.; Gendron-Fitzpatrick, A.; Opitz, J.M. “Double-Muscle” trait in cattle: A possible model for Wiedemann-Beckwith Syndrome. Fetal Pediatr. Pathol. 2006, 25, 9–20. [Google Scholar] [CrossRef]
- Biscarini, F.; Del Corvo, M.; Stella, A.; Alber, A.; Ferencakovic, M.; Pollo, G. Busqueda de las mutaciones causales para artrogriposis y macroglosia en vacuno de raza piemontesa: Resultados preliminares. In Proceedings of the XV Jornadas sobre Producción Animal, Zaragoza, Spain, 14–15 May 2013; Volume II, pp. 538–540. [Google Scholar]
- Beever, J.E.; Marron, B.M. Screening for Arthrogryposis Multiplex in Bovines. U.S. Patent 20,110,151,440 A1, 23 June 2011. [Google Scholar]
- Wiedemar, N.; Riedi, A.K.; Jagannathan, V.; Drögemüller, C.; Meylan, M. Genetic abnormalities in a calf with congenital increased muscular tonus. J. Vet. Intern. Med. 2015, 29, 1418–1421. [Google Scholar] [CrossRef] [Green Version]
- Sartelet, A.; Li, W.; Pailhoux, E.; Richard, C.; Tamma, N.; Karim, L.; Fasquelle, C.; Druet, T.; Coppieters, W.; Georges, M.; et al. Genome-wide next-generation DNA and RNA sequencing reveals a mutation that perturbs splicing of the phosphatidylinositol glycan anchor biosynthesis class H gene (PIGH) and causes arthrogryposis in Belgian Blue cattle. BMC Genom. 2015, 16, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Agerholm, J.S.; McEvoy, F.J.; Menzi, F.; Jagannathan, V.; Drögemüller, C.A. CHRNB1 frameshift mutation is associated with familial arthrogryposis multiplex congenita in Red dairy cattle. BMC Genom. 2016, 17, 479. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Browning, S.R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kijas, J.W.; Hadfield, T.; Naval Sanchez, M.; Cockett, N. Genome-wide association reveals the locus responsible for four-horned ruminant. Anim. Genet. 2016, 47, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrangelo, S.; Sottile, G.; Sardina, M.T.; Sutera, A.M.; Tolone, M.; Di Gerlando, R.; Portolano, B. A combined genome-wide approach identifies a new potential candidate marker associated with the coat color sidedness in cattle. Livest. Sci. 2019, 225, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Sorbolini, S.; Gaspa, G.; Steri, R.; Dimauro, C.; Cellesi, M.; Stella, A.; Marras, G.; Ajmone Marsan, P.; Valentini, A.; Macciotta, N.P.P. Use of canonical discriminant analysis to study signatures of selection in cattle. Genet. Sel. Evol. 2016, 48, 58. [Google Scholar] [CrossRef] [Green Version]
- Aulchencko, Y.S. Effects of Population Structure in Genome-wide Association Studies in Analysis of complex Disease Association Study. In Analysis of Complex Disease Association Studies, 1st ed.; Zeggini, E., Morris, A., Eds.; Accademic Press: London, UK, 2011; Chapter 9; pp. 123–156. ISBN 9780123751430. [Google Scholar]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Cornetti, L.; Tschirren, B. Combining genome-wide association study and FST-based approaches to identify targets of Borrelia-mediated selection in natural rodent hosts. Mol. Ecol. 2020, 29, 386–1397. [Google Scholar] [CrossRef]
- Cesarani, A.; Sechi, T.; Gaspa, G.; Usai, M.G.; Sorbolini, S.; Macciotta, N.P.P.; Carta, A. Investigation of genetic diversity and selection signatures between Sarda and Sardinian Ancestral black, two related sheep breeds with evident morphological differences. Small Rumin. Res. 2019, 177, 68–75. [Google Scholar] [CrossRef]
- Manca, E.; Cesarani, A.; Gaspa, G.; Sorbolini, S.; Macciotta, N.P.; Dimauro, C. Use of the Multivariate Discriminant Analysis for Genome-Wide Association Studies in Cattle. Animals 2020, 10, 1300. [Google Scholar] [CrossRef] [PubMed]
- Windsor, P.A.; Kessell, A.E.; Finnie, J.W. Neurological diseases of ruminant livestock in Australia. V: Congenital neurogenetic disorders of cattle. Aust. Vet. J. 2011, 89, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Sathyanath, R.; Kennedy, T.E. The Netrin Protein Family. Genome Biol. 2009, 10, 239. [Google Scholar] [CrossRef]
- Wang, H.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Tessier-Lavigne, M. Netrin-3, a Mouse Homolog of Human NTN2L, Is Highly Expressed in Sensory Ganglia and Shows Differential Binding to Netrin Receptors. J. Neurosci. 1999, 19, 4938–4947. [Google Scholar] [CrossRef] [Green Version]
- De Castro, M.P.; Aránega, A.; Franco, D. Protein Distribution of Kcnq1, Kcnq2, and Kcnq3 Potassium Channel Subunits during Mouse embryonic Development. Anat. Rec. 2006, 288, 304–315. [Google Scholar] [CrossRef]
- Occhiodoro, T.; Bernheim, L.; Liu, J.H.; Bijlenga, P.; Sinnreich, M.; Bader, C.R.; Fischer-Lougheed, J. Cloning of a Human Ether-a-Go-Go Potassium Channel Expressed in Myoblasts at the Onset of Fusion. FEBS Lett. 1998, 434, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Kakinuma, N.; Wang, Y.; Kiyama, R. Kank Proteins: A New Family of Ankyrin-Repeat Domain-Containing Proteins. BBA-Gen. Subj. 2008, 1780, 128–133. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| SNP | GWAS | Raw CC | ||
|---|---|---|---|---|
| BTA 1 | Disease 2 | Can1 | Can2 | |
| ARS-BFGL-NGS-13673 | 1 | Ma | 0.63 | 0.52 |
| ARS-USDA-AGIL-chr2-17084934-000418 | 2 | Ma | 0.27 | 0.59 |
| ARS-USDA-AGIL-chr4-114395607-000108 | 4 | Ar, Ma | −0.19 | 0.05 |
| ARS-USDA-AGIL-chr7-12174899-000752 | 7 | Ar | −0.18 | −0.14 |
| Hapmap44668-BTA-119022 | 7 | Ar | 0.62 | −0.26 |
| ARS-USDA-AGIL-chr7-18201332-000761 | 7 | Ma | 0.66 | 0.88 |
| ARS-USDA-AGIL-chr8-84099468-000783 | 8 | Ar | −0.15 | −0.72 |
| ARS-USDA-AGIL-chr10-25594159-000176 | 10 | Ar, Ma | −0.37 | 0.15 |
| ARS-USDA-AGIL-chr11-36809347-000009 | 11 | Ar, Ma | −0.16 | 0.22 |
| ARS-BFGL-NGS-15423 | 16 | Ma | 0.02 | 0.02 |
| BovineHD1600007856 | 16 | Ma | 0.37 | 0.25 |
| BovineHD4100012725 | 16 | Ma | 0.72 | −0.11 |
| ARS-USDA-AGIL-chr17-6999864-000318 | 17 | Ma | −0.09 | −0.17 |
| ARS-USDA-AGIL-chr22-32285822-000477 | 22 | Ar, Ma | 0.19 | −0.08 |
| BovineHD2400015279 | 24 | Ar | −0.57 | 0.56 |
| ARS-USDA-AGIL-chr24-25995108-000530 | 24 | Ar, Ma | −0.17 | −0.47 |
| ARS-USDA-AGIL-chr25-1930875-000536 | 25 | Ar | −0.12 | 0.45 |
| BovineHD4100017966 | 26 | Ma | −0.02 | −0.78 |
| BovineHD2600011259 | 26 | Ma | 0.31 | 0.42 |
| BovineHD2600011282 | 26 | Ma | 0.42 | 0.5 |
| BovineHD2600014129 | 26 | Ma | 0.59 | 0.85 |
| BovineHD2800000629 | 28 | Ar | 0.56 | −1.17 |
| ARS-USDA-AGIL-chr29-39842168-000583 | 29 | Ar | −0.40 | −0.65 |
| Within-class average | ||||
| Arthrogryposis | Ar | 1.23 | −1.55 | |
| Macroglossia | Ma | 2.03 | 1.13 | |
| Healthy control | He | −1.62 | 0.16 | |
| BTA | SNP | Gene | |||
|---|---|---|---|---|---|
| Name | Position | Symbol | Name | Location | |
| 4 | ARS-USDA-AGIL-chr4-114395607-000108 | 113,596,650 | KCNH2 | Potassium voltage-gated channel, sub-family H, member 2 | 113,526,185..113,562,025 |
| NOS3 | Nitric oxide synthase 3 | 113,577,075..113,595,527 | |||
| ATG9B | Autophagy-related 9B | 113,594,821..113,605,878 | |||
| ABCB8 | ATP binding cassette subfamily B member 8 | 113,605,775..113,623,178 | |||
| ASIC3 | acid sensing ion channel subunit 3 | 113,624,088..113,629,085 | |||
| 7 | ARS-USDA-AGIL-chr7-12174899-000752 | 11,085,449 | ZNF333 | zinc finger protein 333 | 11,071,151..11,102,144 |
| ADGRE3 | adhesion G protein-coupled receptor E3 | 11,116,555..11,181,632 | |||
| Hapmap44668-BTA-119022 | 85,227,970 | // | no genes in the considered interval | ||
| 8 | ARS-USDA-AGIL-chr8-84099468-000783 | 82,677,581 | ERCC6L2 | ERCC excision repair 6 like 2 | 82,557,942..82,712,671 |
| 10 | ARS-USDA-AGIL-chr10-25594159-000176 | 25,539,231 | OR4E2 | olfactory receptor, family 4, subfamily E, member 2 | 25,539,229..25,540,170 |
| OR10G2 | olfactory receptor, family 10, subfamily G, member 2 | 25,587,007..25,587,963 | |||
| 11 | ARS-USDA-AGIL-chr11-36809347-000009 | 36,957,396 | ACYP2 | acylphosphatase 2 | 36,831,155..37,010,456 |
| TSPYL6 | TSPY like 6 | 36,954,224..36,957,858 | |||
| 22 | ARS-USDA-AGIL-chr22-32285822-000477 | 32,169,050 | FRMD4B | FERM domain containing 4B | 32,022,767..32,382,939 |
| 24 | ARS-USDA-AGIL-chr24-25995108-000530 | 25,684,356 | DSG2 | desmoglein 2 | 25,602,973..25,653,973 |
| DSG3 | desmoglein 3 | 25,666,298..25,699,081 | |||
| DSG4 | desmoglein 4 | 25,725,711..25,755,364 | |||
| BovineHD2400015279 | 53,288,962 | // | no genes in the considered interval | ||
| 25 | ARS-USDA-AGIL-chr25-1930875-000536 | 1,929,340 | CCNF | cyclin F | 1,947,654..1,965,422 |
| TEDC2 | tubulin epsilon and delta complex 2 | 1,966,625..1,970,947 | |||
| NTN3 | netrin 3 | 1,975,804..1,978,415 | |||
| 28 | BovineHD2800000629 | 2,661,658 | // | no genes in the considered interval | |
| 29 | ARS-USDA-AGIL-chr29-39842168-000583 | 39,215,494 | PAG9 | pregnancy-associated glycoprotein 9 | 39,237,412..39,246,707 |
| BTA | SNP | Gene | |||
|---|---|---|---|---|---|
| Name | Position | Symbol | Name | Location | |
| 1 | ARS-BFGL-NGS-13673 | 86,784,250 | // | no genes in the considered interval | |
| 2 | ARS-USDA-AGIL-chr2-17084934-000418 | 17,077,000 | ZNF385B | zinc finger protein 385B | 16,957,646..17,442,243 |
| TRNAC-ACA | transfer RNA cysteine | 17,082,352..17,082,423 | |||
| 4 | ARS-USDA-AGIL-chr4-114395607-000108 | 113,596,650 | KCNH2 | potassium voltage-gated channel, sub-family H, member 2 | 113,526,185..113,562,025 |
| NOS3 | nitric oxide synthase 3 | 113,577,075..113,595,527 | |||
| ATG9B | autophagy related 9B | 113,594,821..113,605,878 | |||
| ABCB8 | ATP binding cassette subfamily B member 8 | 113,605,775..113,623,178 | |||
| ASIC3 | acid-sensing ion channel subunit 3 | 113,624,088..113,629,085 | |||
| 7 | ARS-USDA-AGIL-chr7-18201332-000761 | 16,970,401 | CERS4 | ceramide synthase 4 | 16,897,965..16,933,292 |
| CD320 | CD320 molecule | 16,952,355..16,957,071 | |||
| NDUFA7 | NADH:ubiquinone oxidoreductase subunit A7 | 16,960,703..16,967,936 | |||
| RPS28 | ribosomal protein S28 | 16,968,061..16,969,193 | |||
| KANK3 | KN motif and ankyrin repeat domains 3 | 16,969,360..16,980,814 | |||
| ANGPTL4 | angiopoietin like 4 | 17,005,585..17,012,655 | |||
| 10 | ARS-USDA-AGIL-chr10-25594159-000176 | 25,539,231 | OR4E2 | olfactory receptor, family 4, subfamily E, member 2 | 25,539,229..25,540,170 |
| OR10G2 | olfactory receptor, family 10, subfamily G, member 2 | 25,587,007..25,587,963 | |||
| 11 | ARS-USDA-AGIL-chr11-36809347-000009 | 36,957,396 | ACYP2 | acylphosphatase 2 | 36,831,155..37,010,456 |
| TSPYL6 | TSPY like 6 | 36,954,224..36,957,858 | |||
| 16 | BovineHD1600007856 | 27,494,192 | NVL | nuclear valosin-containing protein-like | 27,322,130..27,502,627 |
| CNIH4 | cornichon family AMPA receptor auxiliary protein 4 | 27,529,034..27,543,276 | |||
| ARS-BFGL-NGS-15423 | 72,258,249 | KCNH1 | potassium voltage-gated channel subfamily H member 1 | 72,205,829..72,642,416 | |
| BovineHD4100012725 | 72,266,300 | ||||
| 17 | ARS-USDA-AGIL-chr17-6999864-000318 | 7,013,884 | LRBA | lipopolysaccharide responsive beige-like anchor protein | 6,799,299..7,556,716 |
| 22 | ARS-USDA-AGIL-chr22-32285822-000477 | 32,169,050 | FRMD4B | FERM domain containing 4B | 32,022,767..32,382,939 |
| 24 | ARS-USDA-AGIL-chr24-25995108-000530 | 25,684,356 | DSG2 | desmoglein 2 | 25,602,973..25,653,973 |
| DSG3 | desmoglein 3 | 25,666,298..25,699,081 | |||
| DSG4 | desmoglein 4 | 25,725,711..25,755,364 | |||
| 26 | BovineHD4100017966 | 40,441,709 | PLPP4 | phospholipid phosphatase 4 | 40,509,729..40,656,785 |
| BovineHD2600011259 | 40,460,199 | ||||
| BovineHD2600011282 | 40,517,972 | ||||
| BovineHD2600014129 | 48,680,201 | // | no genes in the considered interval | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stasio, L.; Albera, A.; Pauciullo, A.; Cesarani, A.; Macciotta, N.P.P.; Gaspa, G. Genetics of Arthrogryposis and Macroglossia in Piemontese Cattle Breed. Animals 2020, 10, 1732. https://doi.org/10.3390/ani10101732
Di Stasio L, Albera A, Pauciullo A, Cesarani A, Macciotta NPP, Gaspa G. Genetics of Arthrogryposis and Macroglossia in Piemontese Cattle Breed. Animals. 2020; 10(10):1732. https://doi.org/10.3390/ani10101732
Chicago/Turabian StyleDi Stasio, Liliana, Andrea Albera, Alfredo Pauciullo, Alberto Cesarani, Nicolò P. P. Macciotta, and Giustino Gaspa. 2020. "Genetics of Arthrogryposis and Macroglossia in Piemontese Cattle Breed" Animals 10, no. 10: 1732. https://doi.org/10.3390/ani10101732
APA StyleDi Stasio, L., Albera, A., Pauciullo, A., Cesarani, A., Macciotta, N. P. P., & Gaspa, G. (2020). Genetics of Arthrogryposis and Macroglossia in Piemontese Cattle Breed. Animals, 10(10), 1732. https://doi.org/10.3390/ani10101732